Predict Gene Expression Part I: Feature Generation#
This is the first of two notebooks which use 6-base data to predict gene expression in mouse embryonic stem cells ES-E14. We aim to use methylation data in a simple set of genomic regions (upstream regions, around TSSs, gene bodies, first exons and introns, other exons and introns, 5 and 3’ UTRs, downstream regions, and CpG islands), and train a gene expression prediction model, using a public dataset of expression. The regions are based on the Gencode annotations, for the mm10 mouse genome.
In this notebook, for each of the regions above, we compute a mean 5mC fraction, a mean 5hmC fraction, a record the number of CpGs in the region (regardless of whether they are methylated or not), and the length of the region. Thus, we capture 4 features per region.
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import pyranges as pr
import seaborn as sns
modality contains a set of functions to extract annotations from GENCODE. Note that GENCODE only provides annotations for the Human and Mouse genomes, and the annotation module of modality only supports the hg38 (GRCh38), mm10 (GRCm38) and mm39 (GRCm39) genomes. In this notebook, we use mm10.
from modality import load_biomodal_dataset
from modality.annotation import (
get_genes,
get_transcription_end_region,
get_tss_region,
get_exons,
get_introns,
get_five_prime_utrs,
get_three_prime_utrs,
get_transcripts,
get_cpg_islands,
)
sns.set_theme()
sns.set_style("whitegrid")
biomodal_palette = ["#003B49", "#9CDBD9", "#F87C56", "#C0DF16", "#05868E"]
sns.set_palette(biomodal_palette)
Load the data#
Our data is stored in a compressed zarr store. modality allows its users to load the zarr store into an object called a ContigDataset, which is a subclass of xarray.Dataset. The ContigDataset gives a useful multidimensional view of our data. In our case, our methylation data (for instance the number of methylated C’s at a given CpG) is represented along two dimensions: the CpG genomic position, and the sample ID.
The function below loads our public mouse dataset for the ES-E14 cell line, using the load_biomodal_dataset function. Note that a user could decide to load their own zarr store instead, using the more general loader method from_zarrz.
The function below does several things:
load the data into a
ContigDatasetcalledds.Drop some variables that we won’t need.
Because the samples are all technical replicates, we sum over all 4 of them with
ds.sumto gain some power.Now that we’ve summed over the samples,
dsis a one-dimensional array along theposdimension. However, internally,modalityoften has to assume thatsample_idis a dimension ofds. So we add it back withds.expand_dims(dim="sample_id", axis=1), and we give it a name withds.assign_coords(sample_id=["sample_0"]).Finally we compute methylation fractions with
ds.assign_fractions.
def load_data(
merge_technical_replicates=True,
merge_cpgs=False,
min_coverage=None,
):
ds = load_biomodal_dataset(
"esE14",
merge_technical_replicates=merge_technical_replicates,
merge_cpgs=merge_cpgs,
)
if min_coverage is not None:
ds = ds.subset_bycoverage(min_coverage=min_coverage)
ds.assign_fractions(
numerators=["num_modc", "num_mc", "num_hmc"],
denominator="num_total_c",
min_coverage=10,
inplace=True,
)
return ds
ds = load_data(
merge_technical_replicates=True,
merge_cpgs=False,
min_coverage=10
)
Below is what our final ContigDataset looks like. It has 26 million CpGs along the pos dimension, and one sample along the sample_id dimension.
print(ds)
<modality.ContigDataset>
<xarray.Dataset> Size: 7GB
Dimensions: (pos: 38787463, sample_id: 1)
Coordinates:
* sample_id (sample_id) <U6 24B 'sample'
contig (pos) <U20 3GB dask.array<chunksize=(80000,), meta=np.ndarray>
ref_position (pos) uint32 155MB dask.array<chunksize=(80000,), meta=np.ndarray>
strand (pos) <U2 310MB dask.array<chunksize=(80000,), meta=np.ndarray>
trinucleotide_context (pos) uint8 39MB dask.array<chunksize=(80000,), meta=np.ndarray>
Dimensions without coordinates: pos
Data variables:
num_c (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_hmc (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_mc (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_modc (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_n (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_other (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_total (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_total_c (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
frac_modc (pos, sample_id) float64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
frac_mc (pos, sample_id) float64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
frac_hmc (pos, sample_id) float64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
Attributes:
context: CG
context_encoding: {'0': 'AAA', '1': 'AAC', '10': 'AGA', '100': 'TAA', '...
contigs: ['chr1', 'chr2', 'chr3', 'chr4', 'chr5', 'chr6', 'chr...
coordinate_basis: 0
description: An evoC dataset of mouse ES-E14 cells, data contains ...
fasta_path: GRCm38p6.fa
input_path: ['CEG1485-EL01-D1115-001.genome.GRCm38p6_primary_asse...
quant_subtype: evoC
ref_name: GRCm38p6
sample_ids: ['CEG1485-EL01-D1115-001', 'CEG1485-EL01-D1115-002', ...
slice_GL456210.1: slice(38760725, 38762983, 1)
slice_GL456211.1: slice(38762983, 38765606, 1)
slice_GL456212.1: slice(38765606, 38766985, 1)
slice_GL456216.1: slice(38766985, 38768808, 1)
slice_GL456221.1: slice(38768808, 38770731, 1)
slice_GL456233.1: slice(38770731, 38773440, 1)
slice_GL456239.1: slice(38773440, 38773885, 1)
slice_GL456354.1: slice(38773885, 38774243, 1)
slice_GL456359.1: slice(38774243, 38774482, 1)
slice_GL456367.1: slice(38774482, 38775051, 1)
slice_GL456368.1: slice(38775051, 38775163, 1)
slice_GL456370.1: slice(38775163, 38775657, 1)
slice_GL456372.1: slice(38775657, 38776011, 1)
slice_GL456378.1: slice(38776011, 38776272, 1)
slice_GL456379.1: slice(38776272, 38776958, 1)
slice_GL456381.1: slice(38776958, 38777177, 1)
slice_GL456382.1: slice(38777177, 38777397, 1)
slice_GL456383.1: slice(38777397, 38778447, 1)
slice_GL456385.1: slice(38778447, 38778745, 1)
slice_GL456387.1: slice(38778745, 38778960, 1)
slice_GL456389.1: slice(38778960, 38779732, 1)
slice_GL456390.1: slice(38779732, 38780195, 1)
slice_GL456392.1: slice(38780195, 38780936, 1)
slice_GL456393.1: slice(38780936, 38781730, 1)
slice_GL456394.1: slice(38781730, 38782021, 1)
slice_GL456396.1: slice(38782021, 38782780, 1)
slice_JH584292.1: slice(38782780, 38782845, 1)
slice_JH584294.1: slice(38782845, 38782849, 1)
slice_JH584295.1: slice(38782849, 38783058, 1)
slice_JH584296.1: slice(38783058, 38783059, 1)
slice_JH584297.1: slice(38783059, 38783079, 1)
slice_JH584299.1: slice(38783079, 38785871, 1)
slice_JH584301.1: slice(38785871, 38785873, 1)
slice_JH584304.1: slice(38785873, 38787463, 1)
slice_chr1: slice(0, 2683651, 1)
slice_chr10: slice(20816493, 22875812, 1)
slice_chr11: slice(22875812, 25073929, 1)
slice_chr12: slice(25073929, 26774508, 1)
slice_chr13: slice(26774508, 28571808, 1)
slice_chr14: slice(28571808, 30195909, 1)
slice_chr15: slice(30195909, 31779607, 1)
slice_chr16: slice(31779607, 33169140, 1)
slice_chr17: slice(33169140, 34705722, 1)
slice_chr18: slice(34705722, 36023233, 1)
slice_chr19: slice(36023233, 37051640, 1)
slice_chr2: slice(2683651, 5512322, 1)
slice_chr3: slice(5512322, 7684834, 1)
slice_chr4: slice(7684834, 10033881, 1)
slice_chr5: slice(10033881, 12510242, 1)
slice_chr6: slice(12510242, 14648397, 1)
slice_chr7: slice(14648397, 16758876, 1)
slice_chr8: slice(16758876, 18817679, 1)
slice_chr9: slice(18817679, 20816493, 1)
slice_chrM: slice(38760292, 38760725, 1)
slice_chrX: slice(37051640, 38619916, 1)
slice_chrY: slice(38619916, 38760292, 1)
zarr_version: 0.1
frac_denominator: num_total_c
frac_min_coverage: 10
Load genes#
Now it is time to start loading genomic annotations. First we load the genes from GENCODE, focusing only on HAVANA protein-coding genes for the mm10 mouse reference genome.
gene_filter = {
"gene_type": "protein_coding",
"source": "HAVANA",
}
genes = get_genes(
reference="mm10",
as_pyranges=True,
filterby=gene_filter,
)
genes = genes.unstrand()
genes.head(5)
| Chromosome | Source | Type | Start | End | Score | Phase | Id | Gene_id | Gene_type | Gene_name | Level | Mgi_id | Havana_gene | Tag | Ranges_ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1 | HAVANA | gene | 4807787 | 4848409 | . | . | ENSMUSG00000025903.14 | ENSMUSG00000025903.14 | protein_coding | Lypla1 | 2 | MGI:1344588 | OTTMUSG00000021562.4 | overlapping_locus | 0 |
| 1 | chr1 | HAVANA | gene | 4807891 | 4886769 | . | . | ENSMUSG00000104217.1 | ENSMUSG00000104217.1 | protein_coding | Gm37988 | 2 | MGI:5611216 | OTTMUSG00000050100.1 | overlapping_locus | 1 |
| 2 | chr1 | HAVANA | gene | 4857813 | 4897908 | . | . | ENSMUSG00000033813.15 | ENSMUSG00000033813.15 | protein_coding | Tcea1 | 2 | MGI:1196624 | OTTMUSG00000042348.1 | overlapping_locus | 2 |
| 3 | chr1 | HAVANA | gene | 5070017 | 5162528 | . | . | ENSMUSG00000033793.12 | ENSMUSG00000033793.12 | protein_coding | Atp6v1h | 2 | MGI:1914864 | OTTMUSG00000050145.9 | 3 | |
| 4 | chr1 | HAVANA | gene | 5588465 | 5606130 | . | . | ENSMUSG00000025905.14 | ENSMUSG00000025905.14 | protein_coding | Oprk1 | 2 | MGI:97439 | OTTMUSG00000034734.3 | 4 |
Identify relevant transcript for each gene#
For each gene, we also need to extract the most relevant transcript. This is because some of the genomic annotations we’ll want to extract later are defined at a per-transcript level instead of a per-gene level (exons, introns, 5’ UTRs, and 3’ UTRs).
transcripts = get_transcripts(
reference="mm10",
contig=None,
start=None,
end=None,
as_pyranges=False,
)
def select_transcript_based_on_tag(df):
# for each transcript in df, select the one with the highest priority tag
# priorities are:
# 1. 'basic,appris_principal_1,CCDS'
# 2. 'basic,appris_principal_1'
# 3. 'basic,CCDS'
# 4. 'basic'
# but with 'exp_conf' (experimentally confirmed) tag, the priority is higher.
priorties = {
'basic,appris_principal_1,exp_conf,CCDS': 1,
'basic,appris_principal_1,CCDS': 1,
'basic,appris_principal_1,exp_conf': 3,
'basic,appris_principal_1': 4,
'basic,exp_conf,CCDS': 5,
'basic,CCDS': 6,
'basic,exp_conf': 7,
'basic': 8
}
# sort the dataframe by the priority of the tags
df['tag_priority'] = df.tag.map(priorties)
df = df.sort_values(by='tag_priority')
# drop duplicates, keeping the first one
df = df.drop_duplicates(subset='gene_id', keep='first')
return df[["gene_id", "transcript_id"]]
selected_transcripts = transcripts.groupby('gene_id').apply(
select_transcript_based_on_tag
).reset_index(drop=True)
selected_transcripts.head()
| gene_id | transcript_id | |
|---|---|---|
| 0 | ENSMUSG00000000001.4 | ENSMUST00000000001.4 |
| 1 | ENSMUSG00000000003.15 | ENSMUST00000000003.13 |
| 2 | ENSMUSG00000000028.15 | ENSMUST00000000028.13 |
| 3 | ENSMUSG00000000037.17 | ENSMUST00000101113.8 |
| 4 | ENSMUSG00000000049.11 | ENSMUST00000000049.5 |
Get the upstream region#
We define the upstream region as the region 2kb upstream of the TSS, split into subregions. This region is important as it is likely to contain the promoter.
There is some granularity in the upstream region, which we don’t want to wash out by summarising over a region that would be too large. In particular, promoters are small regions located upstream of the gene, which play a fundamental role in transcription. They may be core promoters very close to the TSS, proximal promoters within a few hundred bp, or more distal promoters, further from the TSS.
Here we’re going to use modality’s get_tss function, which contains a start_offset argument. A negative value, for instance -500, means that we start from 500bp upstream of the TSS. The span argument determines the length of the region we are interested. So start_offset=-500 and span=250 will grab the region from 500 to 250bp upstream of the TSS. Here we include 5 regions, from 2kb to 1.5kb, from 1.5kb to 1kb, from 1kb to 500bp, from 500bp to 250bp, and from 250bp to 0 (the TSS). We chose these to get a bit more granularity near the TSS.
distances = [-2000, -1500, -1000, -500, -250]
spans = np.diff(distances+[0])
default_args = {
"contig": None,
"start": None,
"end": None,
"reference": "mm10",
"as_pyranges": True,
"protein_coding": True,
"filterby": None,
}
upstream_dict = {}
for distance, span in zip(distances, spans):
upstream = get_tss_region(
start_offset=distance,
span=span,
**default_args,
)
upstream = upstream.unstrand()
upstream_dict[f"upstream_{-distance}"] = upstream
upstream_dict["upstream_250"].head()
| Chromosome | Source | Type | Start | End | Score | Phase | Id | Gene_id | Gene_type | Gene_name | Level | Mgi_id | Havana_gene | Tag | Ranges_ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1 | HAVANA | gene | 4807537 | 4807787 | . | . | ENSMUSG00000025903.14 | ENSMUSG00000025903.14 | protein_coding | Lypla1 | 2 | MGI:1344588 | OTTMUSG00000021562.4 | overlapping_locus | 0 |
| 1 | chr1 | HAVANA | gene | 4807641 | 4807891 | . | . | ENSMUSG00000104217.1 | ENSMUSG00000104217.1 | protein_coding | Gm37988 | 2 | MGI:5611216 | OTTMUSG00000050100.1 | overlapping_locus | 1 |
| 2 | chr1 | HAVANA | gene | 4857563 | 4857813 | . | . | ENSMUSG00000033813.15 | ENSMUSG00000033813.15 | protein_coding | Tcea1 | 2 | MGI:1196624 | OTTMUSG00000042348.1 | overlapping_locus | 2 |
| 3 | chr1 | HAVANA | gene | 5069767 | 5070017 | . | . | ENSMUSG00000033793.12 | ENSMUSG00000033793.12 | protein_coding | Atp6v1h | 2 | MGI:1914864 | OTTMUSG00000050145.9 | 3 | |
| 4 | chr1 | HAVANA | gene | 5588215 | 5588465 | . | . | ENSMUSG00000025905.14 | ENSMUSG00000025905.14 | protein_coding | Oprk1 | 2 | MGI:97439 | OTTMUSG00000034734.3 | 4 | |
| 5 | chr1 | HAVANA | gene | 6205946 | 6206196 | . | . | ENSMUSG00000025907.14 | ENSMUSG00000025907.14 | protein_coding | Rb1cc1 | 2 | MGI:1341850 | OTTMUSG00000033467.12 | 5 | |
| 6 | chr1 | HAVANA | gene | 6358967 | 6359217 | . | . | ENSMUSG00000087247.3 | ENSMUSG00000087247.3 | protein_coding | Alkal1 | 2 | MGI:3645495 | OTTMUSG00000050239.2 | overlapping_locus | 6 |
| 7 | chr1 | HAVANA | gene | 6486980 | 6487230 | . | . | ENSMUSG00000033740.17 | ENSMUSG00000033740.17 | protein_coding | St18 | 1 | MGI:2446700 | OTTMUSG00000024833.6 | 7 |
Get the region around TSS#
We define the TSS region as 250bp each side of the TSS. Note that this region will overlap with the upstream region above, but also capture relevant information for 250bp downstream of the TSS, inside the gene region.
around_tss = get_tss_region(
start_offset=-200,
span=400,
**default_args,
)
around_tss = around_tss.unstrand()
Get the downstream region#
We define the downstream region as a series of subregions of 1kb length, downstream of the gene body.
distances = np.arange(0, 5000, 1000)
downstream_dict = {}
for distance in distances:
downstream = get_transcription_end_region(
span=1000,
start_offset=distance,
**default_args,
)
downstream = downstream.unstrand()
downstream_dict[f"downstream_{distance}"] = downstream
Get exons#
Next we grab all the exons. This will return all the exons for all the genes in the GENCODE annotation. But in a previous cell, we have identified a list of selected transcripts, so we subset our exons to that list of transcripts.
exons = get_exons(reference="mm10")
exons = exons[exons.Transcript_id.isin(selected_transcripts.transcript_id)]
Get the first exon#
Exons in GENCODE have an Exon_number field which gives the position of the exon in the transcript from its 5’ end. Therefore we can directly pull out the first exon of each transcript as a standalone annotation.
first_exons = exons[exons.Exon_number == "1"]
Now that we have the first exon, we can remove it from the other exons feature.
exons = exons[exons.Exon_number.astype("int") > 1]
Get introns#
Introns are not readily available in GENCODE annotations, but we can use modality’s get_introns function to collect these. This function collects all exons, 5’ UTRS, 3’UTRs and groups them by transcript ID. It then matches the transcript ID with a gene ID, and grabs that gene from the gene list. Finally it subtracts the exons, 5’ UTRS and 3’UTRs from the gene, leaving a set of introns per transcript.
introns = get_introns(
reference="mm10",
transcripts=selected_transcripts.transcript_id.values,
nb_workers=8,
)
Get the first intron#
Similarly to the first exon, we can also grab the first intron, as get_introns() returns a field with the order in which the introns appear from the 5’ end.
first_introns = introns[introns.Intron_number == "1"]
Now that we have the first intron, we can remove it from the other intron feature.
introns = introns[introns.Intron_number.astype("int") > 1]
first_introns.head()
| Transcript_id | Level_1 | Chromosome | Source | Type | Start | End | Score | Strand | Phase | ... | Gene_id | Gene_type | Gene_name | Level | Mgi_id | Havana_gene | Tag | Ranges_id | Intron_number | Ranges_ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ENSMUST00000000514.10 | 0 | chr1 | HAVANA | intron | 107590286 | 107597459 | . | + | . | ... | ENSMUSG00000026315.13 | protein_coding | Serpinb8 | 2 | MGI:894657 | OTTMUSG00000020994.2 | 317.0 | 1 | 0 | |
| 1 | ENSMUST00000001027.6 | 0 | chr1 | HAVANA | intron | 58030057 | 58041435 | . | + | . | ... | ENSMUSG00000063558.4 | protein_coding | Aox1 | 2 | MGI:88035 | OTTMUSG00000029784.3 | 117.0 | 1 | 7 | |
| 2 | ENSMUST00000001166.13 | 0 | chr1 | HAVANA | intron | 36513152 | 36518951 | . | + | . | ... | ENSMUSG00000001138.13 | protein_coding | Cnnm3 | 2 | MGI:2151055 | OTTMUSG00000021763.2 | 57.0 | 1 | 41 | |
| 3 | ENSMUST00000004829.12 | 0 | chr1 | HAVANA | intron | 171559384 | 171573680 | . | + | . | ... | ENSMUSG00000004709.14 | protein_coding | Cd244a | 2 | MGI:109294 | OTTMUSG00000016914.2 | 510.0 | 1 | 48 | |
| 4 | ENSMUST00000005824.11 | 0 | chr1 | HAVANA | intron | 171225053 | 171225081 | . | + | . | ... | ENSMUSG00000005681.12 | protein_coding | Apoa2 | 2 | MGI:88050 | OTTMUSG00000021666.2 | 499.0 | 1 | 57 | |
| 5 | ENSMUST00000005907.11 | 0 | chr1 | HAVANA | intron | 165788680 | 165788688 | . | + | . | ... | ENSMUSG00000005763.15 | protein_coding | Cd247 | 2 | MGI:88334 | OTTMUSG00000034863.5 | overlapping_locus | 480.0 | 1 | 62 |
| 6 | ENSMUST00000006716.7 | 0 | chr1 | HAVANA | intron | 74772221 | 74782101 | . | + | . | ... | ENSMUSG00000033227.7 | protein_coding | Wnt6 | 2 | MGI:98960 | OTTMUSG00000048253.1 | 179.0 | 1 | 71 | |
| 7 | ENSMUST00000006718.14 | 0 | chr1 | HAVANA | intron | 74792282 | 74793362 | . | + | . | ... | ENSMUSG00000026167.14 | protein_coding | Wnt10a | 2 | MGI:108071 | OTTMUSG00000020879.2 | 180.0 | 1 | 74 |
8 rows × 21 columns
Get 5’ UTRs#
We also grab all the 5’ UTRs that are in the transcripts of interest.
five_prime_utrs = get_five_prime_utrs(reference="mm10")
five_prime_utrs = five_prime_utrs[
five_prime_utrs.Transcript_id.isin(selected_transcripts.transcript_id)
]
Get 3’ UTR#
And we do the same for all the 3’ UTRs that are in the transcripts of interest.
three_prime_utrs = get_three_prime_utrs(reference="mm10")
three_prime_utrs = three_prime_utrs[
three_prime_utrs.Transcript_id.isin(selected_transcripts.transcript_id)
]
Get CpG Islands#
CpG islands are regions that contain an elevated number of CpGs and tend to have low overall methylation. They are often associated with promoters and can regulate gene expression (see, e.g., Deaton & Bird 2011). modality has a get_cpg_islands which gets CpG islands from the UCSC Genome Browser.
Note that when loading the mouse CpG islands from UCSC, the chromosomes are called “chr1”, “chr2”, etc…, while GENCODE mouse chromosomes are called “1”, “2”, etc…, so we need a quick function to fix that.
def modify_chrom_series(df):
df.Chromosome = df.Chromosome.apply(lambda val: val.replace("chr", ""))
return df
def fix_chrom(regions):
return regions.apply(modify_chrom_series)
cpg_islands = get_cpg_islands(reference="mm10")
# cpg_islands = fix_chrom(cpg_islands)
cpg_islands.head()
| Index | Bin | Chromosome | Start | End | Length | Cpgnum | Gcnum | Percpg | Pergc | Obsexp | Type | Ranges_ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 4 | 611 | chr1 | 3531624 | 3531843 | 219 | 27 | 167 | 24.7 | 76.3 | 0.86 | cpg_island | 0 |
| 1 | 5 | 613 | chr1 | 3670619 | 3671074 | 455 | 34 | 293 | 14.9 | 64.4 | 0.72 | cpg_island | 1 |
| 2 | 6 | 613 | chr1 | 3671654 | 3672156 | 502 | 45 | 348 | 17.9 | 69.3 | 0.75 | cpg_island | 2 |
| 3 | 7 | 619 | chr1 | 4491701 | 4493673 | 1972 | 165 | 1286 | 16.7 | 65.2 | 0.79 | cpg_island | 3 |
| 4 | 8 | 619 | chr1 | 4496947 | 4497608 | 661 | 47 | 393 | 14.2 | 59.5 | 0.81 | cpg_island | 4 |
| 5 | 9 | 619 | chr1 | 4571641 | 4572075 | 434 | 44 | 265 | 20.3 | 61.1 | 1.09 | cpg_island | 5 |
| 6 | 10 | 620 | chr1 | 4689184 | 4689397 | 213 | 24 | 149 | 22.5 | 70.0 | 0.96 | cpg_island | 6 |
| 7 | 11 | 621 | chr1 | 4785376 | 4785814 | 438 | 49 | 289 | 22.4 | 66.0 | 1.04 | cpg_island | 7 |
Now that we have the CpG islands, we need to match them to genes. Here, as a first approximation, we use pyranges.nearest to find the CpG island nearest to each gene.
cpg_islands_by_genes = genes.nearest(cpg_islands, suffix="_island")
cpg_islands_by_genes.Start = cpg_islands_by_genes.Start_island
cpg_islands_by_genes.End = cpg_islands_by_genes.End_island
cpg_islands_by_genes
| Chromosome | Source | Type | Start | End | Score | Phase | Id | Gene_id | Gene_type | ... | End_island | Length | Cpgnum | Gcnum | Percpg | Pergc | Obsexp | Type_island | Ranges_ID_island | Distance | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1 | HAVANA | gene | 4807559 | 4808103 | . | . | ENSMUSG00000025903.14 | ENSMUSG00000025903.14 | protein_coding | ... | 4808103 | 544 | 73 | 367 | 26.8 | 67.5 | 1.18 | cpg_island | 8 | 0 |
| 1 | chr1 | HAVANA | gene | 4807559 | 4808103 | . | . | ENSMUSG00000104217.1 | ENSMUSG00000104217.1 | protein_coding | ... | 4808103 | 544 | 73 | 367 | 26.8 | 67.5 | 1.18 | cpg_island | 8 | 0 |
| 2 | chr1 | HAVANA | gene | 4857465 | 4858372 | . | . | ENSMUSG00000033813.15 | ENSMUSG00000033813.15 | protein_coding | ... | 4858372 | 907 | 83 | 545 | 18.3 | 60.1 | 1.01 | cpg_island | 9 | 0 |
| 3 | chr1 | HAVANA | gene | 5083039 | 5083536 | . | . | ENSMUSG00000033793.12 | ENSMUSG00000033793.12 | protein_coding | ... | 5083536 | 497 | 49 | 329 | 19.7 | 66.2 | 0.90 | cpg_island | 11 | 0 |
| 4 | chr1 | HAVANA | gene | 6214430 | 6215332 | . | . | ENSMUSG00000025907.14 | ENSMUSG00000025907.14 | protein_coding | ... | 6215332 | 902 | 107 | 645 | 23.7 | 71.5 | 0.93 | cpg_island | 13 | 0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 21668 | chrY | HAVANA | gene | 90741077 | 90744975 | . | . | ENSMUSG00000099856.1 | ENSMUSG00000099856.1 | protein_coding | ... | 90744975 | 3898 | 251 | 2210 | 12.9 | 56.7 | 0.84 | cpg_island | 15998 | 3165388 |
| 21669 | chrY | HAVANA | gene | 90741077 | 90744975 | . | . | ENSMUSG00000101915.1 | ENSMUSG00000101915.1 | protein_coding | ... | 90744975 | 3898 | 251 | 2210 | 12.9 | 56.7 | 0.84 | cpg_island | 15998 | 2661585 |
| 21670 | chrY | HAVANA | gene | 90741077 | 90744975 | . | . | ENSMUSG00000102045.1 | ENSMUSG00000102045.1 | protein_coding | ... | 90744975 | 3898 | 251 | 2210 | 12.9 | 56.7 | 0.84 | cpg_island | 15998 | 1662064 |
| 21671 | chrY | HAVANA | gene | 90741077 | 90744975 | . | . | ENSMUSG00000100608.1 | ENSMUSG00000100608.1 | protein_coding | ... | 90744975 | 3898 | 251 | 2210 | 12.9 | 56.7 | 0.84 | cpg_island | 15998 | 995547 |
| 21672 | chrY | HAVANA | gene | 90741077 | 90744975 | . | . | ENSMUSG00000096178.7 | ENSMUSG00000096178.7 | protein_coding | ... | 90744975 | 3898 | 251 | 2210 | 12.9 | 56.7 | 0.84 | cpg_island | 15998 | 307816 |
21673 rows × 29 columns
Create features with reduce_byranges#
We use a method of modality.ContigDataset called reduce_byranges which allows to reduce our contig dataset to summarise methylation information over a list of genomic ranges (in our case, the ranges that we created above). These ranges should be in the form of pyranges objects. In principle, a user could also pass their own ranges, for instance reading them from a bed or gff file - see pyranges documentation.
regions_dict = upstream_dict.copy()
regions_dict.update(
{
"around_tss": around_tss,
"genes": genes,
"first_exons": first_exons,
"first_introns": first_introns,
"cpg_islands": cpg_islands_by_genes,
}
)
regions_dict.update(downstream_dict)
to_drop = ["Score", "Source", "Phase", "Type"]
for region in regions_dict:
regions_dict[region].Region = region
try:
regions_dict[region] = regions_dict[region].drop(to_drop)
except:
pass
list(regions_dict.values())
[+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4805787 | 4806287 | ENSMUSG00000025903.14 | ... |
| chr1 | 4805891 | 4806391 | ENSMUSG00000104217.1 | ... |
| chr1 | 4855813 | 4856313 | ENSMUSG00000033813.15 | ... |
| chr1 | 5068017 | 5068517 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79152621 | 79153121 | ENSMUSG00000095867.2 | ... |
| chrY | 84566406 | 84566906 | ENSMUSG00000094660.2 | ... |
| chrY | 85532407 | 85532907 | ENSMUSG00000095650.2 | ... |
| chrY | 89747031 | 89747531 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4806287 | 4806787 | ENSMUSG00000025903.14 | ... |
| chr1 | 4806391 | 4806891 | ENSMUSG00000104217.1 | ... |
| chr1 | 4856313 | 4856813 | ENSMUSG00000033813.15 | ... |
| chr1 | 5068517 | 5069017 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79152121 | 79152621 | ENSMUSG00000095867.2 | ... |
| chrY | 84565906 | 84566406 | ENSMUSG00000094660.2 | ... |
| chrY | 85531907 | 85532407 | ENSMUSG00000095650.2 | ... |
| chrY | 89746531 | 89747031 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4806787 | 4807287 | ENSMUSG00000025903.14 | ... |
| chr1 | 4806891 | 4807391 | ENSMUSG00000104217.1 | ... |
| chr1 | 4856813 | 4857313 | ENSMUSG00000033813.15 | ... |
| chr1 | 5069017 | 5069517 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79151621 | 79152121 | ENSMUSG00000095867.2 | ... |
| chrY | 84565406 | 84565906 | ENSMUSG00000094660.2 | ... |
| chrY | 85531407 | 85531907 | ENSMUSG00000095650.2 | ... |
| chrY | 89746031 | 89746531 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4807287 | 4807537 | ENSMUSG00000025903.14 | ... |
| chr1 | 4807391 | 4807641 | ENSMUSG00000104217.1 | ... |
| chr1 | 4857313 | 4857563 | ENSMUSG00000033813.15 | ... |
| chr1 | 5069517 | 5069767 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79151371 | 79151621 | ENSMUSG00000095867.2 | ... |
| chrY | 84565156 | 84565406 | ENSMUSG00000094660.2 | ... |
| chrY | 85531157 | 85531407 | ENSMUSG00000095650.2 | ... |
| chrY | 89745781 | 89746031 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4807537 | 4807787 | ENSMUSG00000025903.14 | ... |
| chr1 | 4807641 | 4807891 | ENSMUSG00000104217.1 | ... |
| chr1 | 4857563 | 4857813 | ENSMUSG00000033813.15 | ... |
| chr1 | 5069767 | 5070017 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79151121 | 79151371 | ENSMUSG00000095867.2 | ... |
| chrY | 84564906 | 84565156 | ENSMUSG00000094660.2 | ... |
| chrY | 85530907 | 85531157 | ENSMUSG00000095650.2 | ... |
| chrY | 89745531 | 89745781 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4807587 | 4807987 | ENSMUSG00000025903.14 | ... |
| chr1 | 4807691 | 4808091 | ENSMUSG00000104217.1 | ... |
| chr1 | 4857613 | 4858013 | ENSMUSG00000033813.15 | ... |
| chr1 | 5069817 | 5070217 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79150921 | 79151321 | ENSMUSG00000095867.2 | ... |
| chrY | 84564706 | 84565106 | ENSMUSG00000094660.2 | ... |
| chrY | 85530707 | 85531107 | ENSMUSG00000095650.2 | ... |
| chrY | 89745331 | 89745731 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4807787 | 4848409 | ENSMUSG00000025903.14 | ... |
| chr1 | 4807891 | 4886769 | ENSMUSG00000104217.1 | ... |
| chr1 | 4857813 | 4897908 | ENSMUSG00000033813.15 | ... |
| chr1 | 5070017 | 5162528 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79148788 | 79151121 | ENSMUSG00000095867.2 | ... |
| chrY | 84562571 | 84564906 | ENSMUSG00000094660.2 | ... |
| chrY | 85528516 | 85530907 | ENSMUSG00000095650.2 | ... |
| chrY | 89713423 | 89745531 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+--------------+-------+
| Chromosome | Start | End | Strand | +21 |
| (category) | (int64) | (int64) | (category) | ... |
|--------------+-----------+-----------+--------------+-------|
| chr1 | 4807822 | 4807981 | + | ... |
| chr1 | 4807891 | 4807981 | + | ... |
| chr1 | 4857813 | 4857975 | + | ... |
| chr1 | 5070017 | 5070143 | + | ... |
| ... | ... | ... | ... | ... |
| chrY | 79151002 | 79151121 | - | ... |
| chrY | 84564788 | 84564906 | - | ... |
| chrY | 85530854 | 85530907 | - | ... |
| chrY | 89745488 | 89745531 | - | ... |
+--------------+-----------+-----------+--------------+-------+
Stranded PyRanges object has 21,541 rows and 25 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome and Strand.
21 hidden columns: Id, Parent, Gene_id, Transcript_id, Gene_type, ... (+ 16 more.),
+-----------------------+-----------+--------------+-----------+-------+
| Transcript_id | Level_1 | Chromosome | Start | +14 |
| (object) | (int64) | (category) | (int64) | ... |
|-----------------------+-----------+--------------+-----------+-------|
| ENSMUST00000000514.10 | 0 | chr1 | 107590286 | ... |
| ENSMUST00000001027.6 | 0 | chr1 | 58030057 | ... |
| ENSMUST00000001166.13 | 0 | chr1 | 36513152 | ... |
| ENSMUST00000004829.12 | 0 | chr1 | 171559384 | ... |
| ... | ... | ... | ... | ... |
| ENSMUST00000191443.6 | 8 | chrY | 40315473 | ... |
| ENSMUST00000191675.1 | 8 | chrY | 20992252 | ... |
| ENSMUST00000192521.1 | 8 | chrY | 22049944 | ... |
| ENSMUST00000192892.1 | 8 | chrY | 22859847 | ... |
+-----------------------+-----------+--------------+-----------+-------+
Stranded PyRanges object has 20,398 rows and 18 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome and Strand.
14 hidden columns: End, Strand, Id, Gene_id, Gene_type, Gene_name, Level, Mgi_id, ... (+ 6 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +22 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4807559 | 4808103 | ENSMUSG00000025903.14 | ... |
| chr1 | 4807559 | 4808103 | ENSMUSG00000104217.1 | ... |
| chr1 | 4857465 | 4858372 | ENSMUSG00000033813.15 | ... |
| chr1 | 5083039 | 5083536 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 90741077 | 90744975 | ENSMUSG00000101915.1 | ... |
| chrY | 90741077 | 90744975 | ENSMUSG00000102045.1 | ... |
| chrY | 90741077 | 90744975 | ENSMUSG00000100608.1 | ... |
| chrY | 90741077 | 90744975 | ENSMUSG00000096178.7 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 26 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
22 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 16 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4848409 | 4849409 | ENSMUSG00000025903.14 | ... |
| chr1 | 4886769 | 4887769 | ENSMUSG00000104217.1 | ... |
| chr1 | 4897908 | 4898908 | ENSMUSG00000033813.15 | ... |
| chr1 | 5162528 | 5163528 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79147788 | 79148788 | ENSMUSG00000095867.2 | ... |
| chrY | 84561571 | 84562571 | ENSMUSG00000094660.2 | ... |
| chrY | 85527516 | 85528516 | ENSMUSG00000095650.2 | ... |
| chrY | 89712423 | 89713423 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4849409 | 4850409 | ENSMUSG00000025903.14 | ... |
| chr1 | 4887769 | 4888769 | ENSMUSG00000104217.1 | ... |
| chr1 | 4898908 | 4899908 | ENSMUSG00000033813.15 | ... |
| chr1 | 5163528 | 5164528 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79146788 | 79147788 | ENSMUSG00000095867.2 | ... |
| chrY | 84560571 | 84561571 | ENSMUSG00000094660.2 | ... |
| chrY | 85526516 | 85527516 | ENSMUSG00000095650.2 | ... |
| chrY | 89711423 | 89712423 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4850409 | 4851409 | ENSMUSG00000025903.14 | ... |
| chr1 | 4888769 | 4889769 | ENSMUSG00000104217.1 | ... |
| chr1 | 4899908 | 4900908 | ENSMUSG00000033813.15 | ... |
| chr1 | 5164528 | 5165528 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79145788 | 79146788 | ENSMUSG00000095867.2 | ... |
| chrY | 84559571 | 84560571 | ENSMUSG00000094660.2 | ... |
| chrY | 85525516 | 85526516 | ENSMUSG00000095650.2 | ... |
| chrY | 89710423 | 89711423 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4851409 | 4852409 | ENSMUSG00000025903.14 | ... |
| chr1 | 4889769 | 4890769 | ENSMUSG00000104217.1 | ... |
| chr1 | 4900908 | 4901908 | ENSMUSG00000033813.15 | ... |
| chr1 | 5165528 | 5166528 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79144788 | 79145788 | ENSMUSG00000095867.2 | ... |
| chrY | 84558571 | 84559571 | ENSMUSG00000094660.2 | ... |
| chrY | 85524516 | 85525516 | ENSMUSG00000095650.2 | ... |
| chrY | 89709423 | 89710423 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.),
+--------------+-----------+-----------+-----------------------+-------+
| Chromosome | Start | End | Id | +9 |
| (category) | (int64) | (int64) | (object) | ... |
|--------------+-----------+-----------+-----------------------+-------|
| chr1 | 4852409 | 4853409 | ENSMUSG00000025903.14 | ... |
| chr1 | 4890769 | 4891769 | ENSMUSG00000104217.1 | ... |
| chr1 | 4901908 | 4902908 | ENSMUSG00000033813.15 | ... |
| chr1 | 5166528 | 5167528 | ENSMUSG00000033793.12 | ... |
| ... | ... | ... | ... | ... |
| chrY | 79143788 | 79144788 | ENSMUSG00000095867.2 | ... |
| chrY | 84557571 | 84558571 | ENSMUSG00000094660.2 | ... |
| chrY | 85523516 | 85524516 | ENSMUSG00000095650.2 | ... |
| chrY | 89708423 | 89709423 | ENSMUSG00000100608.1 | ... |
+--------------+-----------+-----------+-----------------------+-------+
Unstranded PyRanges object has 21,673 rows and 13 columns from 21 chromosomes.
For printing, the PyRanges was sorted on Chromosome.
9 hidden columns: Gene_id, Gene_type, Gene_name, Level, Mgi_id, Havana_gene, ... (+ 3 more.)]
list(regions_dict.values())[0][["Chromosome", "Start", "End"]].head()
| Chromosome | Start | End | |
|---|---|---|---|
| 0 | chr1 | 4805787 | 4806287 |
| 1 | chr1 | 4805891 | 4806391 |
| 2 | chr1 | 4855813 | 4856313 |
| 3 | chr1 | 5068017 | 5068517 |
| 4 | chr1 | 5586465 | 5586965 |
| 5 | chr1 | 6204196 | 6204696 |
| 6 | chr1 | 6357217 | 6357717 |
| 7 | chr1 | 6485230 | 6485730 |
len(list(regions_dict.values()))
15
for r in list(regions_dict.values()):
print(len(r))
21673
21673
21673
21673
21673
21673
21673
21541
20398
21673
21673
21673
21673
21673
21673
ds
<xarray.Dataset> Size: 7GB
Dimensions: (pos: 38787463, sample_id: 1)
Coordinates:
* sample_id (sample_id) <U6 24B 'sample'
contig (pos) <U20 3GB dask.array<chunksize=(80000,), meta=np.ndarray>
ref_position (pos) uint32 155MB dask.array<chunksize=(80000,), meta=np.ndarray>
strand (pos) <U2 310MB dask.array<chunksize=(80000,), meta=np.ndarray>
trinucleotide_context (pos) uint8 39MB dask.array<chunksize=(80000,), meta=np.ndarray>
Dimensions without coordinates: pos
Data variables:
num_c (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_hmc (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_mc (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_modc (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_n (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_other (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_total (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
num_total_c (pos, sample_id) uint64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
frac_modc (pos, sample_id) float64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
frac_mc (pos, sample_id) float64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
frac_hmc (pos, sample_id) float64 310MB dask.array<chunksize=(80000, 1), meta=np.ndarray>
Attributes:
context: CG
context_encoding: {'0': 'AAA', '1': 'AAC', '10': 'AGA', '100': 'TAA', '...
contigs: ['chr1', 'chr2', 'chr3', 'chr4', 'chr5', 'chr6', 'chr...
coordinate_basis: 0
description: An evoC dataset of mouse ES-E14 cells, data contains ...
fasta_path: GRCm38p6.fa
input_path: ['CEG1485-EL01-D1115-001.genome.GRCm38p6_primary_asse...
quant_subtype: evoC
ref_name: GRCm38p6
sample_ids: ['CEG1485-EL01-D1115-001', 'CEG1485-EL01-D1115-002', ...
slice_GL456210.1: slice(38760725, 38762983, 1)
slice_GL456211.1: slice(38762983, 38765606, 1)
slice_GL456212.1: slice(38765606, 38766985, 1)
slice_GL456216.1: slice(38766985, 38768808, 1)
slice_GL456221.1: slice(38768808, 38770731, 1)
slice_GL456233.1: slice(38770731, 38773440, 1)
slice_GL456239.1: slice(38773440, 38773885, 1)
slice_GL456354.1: slice(38773885, 38774243, 1)
slice_GL456359.1: slice(38774243, 38774482, 1)
slice_GL456367.1: slice(38774482, 38775051, 1)
slice_GL456368.1: slice(38775051, 38775163, 1)
slice_GL456370.1: slice(38775163, 38775657, 1)
slice_GL456372.1: slice(38775657, 38776011, 1)
slice_GL456378.1: slice(38776011, 38776272, 1)
slice_GL456379.1: slice(38776272, 38776958, 1)
slice_GL456381.1: slice(38776958, 38777177, 1)
slice_GL456382.1: slice(38777177, 38777397, 1)
slice_GL456383.1: slice(38777397, 38778447, 1)
slice_GL456385.1: slice(38778447, 38778745, 1)
slice_GL456387.1: slice(38778745, 38778960, 1)
slice_GL456389.1: slice(38778960, 38779732, 1)
slice_GL456390.1: slice(38779732, 38780195, 1)
slice_GL456392.1: slice(38780195, 38780936, 1)
slice_GL456393.1: slice(38780936, 38781730, 1)
slice_GL456394.1: slice(38781730, 38782021, 1)
slice_GL456396.1: slice(38782021, 38782780, 1)
slice_JH584292.1: slice(38782780, 38782845, 1)
slice_JH584294.1: slice(38782845, 38782849, 1)
slice_JH584295.1: slice(38782849, 38783058, 1)
slice_JH584296.1: slice(38783058, 38783059, 1)
slice_JH584297.1: slice(38783059, 38783079, 1)
slice_JH584299.1: slice(38783079, 38785871, 1)
slice_JH584301.1: slice(38785871, 38785873, 1)
slice_JH584304.1: slice(38785873, 38787463, 1)
slice_chr1: slice(0, 2683651, 1)
slice_chr10: slice(20816493, 22875812, 1)
slice_chr11: slice(22875812, 25073929, 1)
slice_chr12: slice(25073929, 26774508, 1)
slice_chr13: slice(26774508, 28571808, 1)
slice_chr14: slice(28571808, 30195909, 1)
slice_chr15: slice(30195909, 31779607, 1)
slice_chr16: slice(31779607, 33169140, 1)
slice_chr17: slice(33169140, 34705722, 1)
slice_chr18: slice(34705722, 36023233, 1)
slice_chr19: slice(36023233, 37051640, 1)
slice_chr2: slice(2683651, 5512322, 1)
slice_chr3: slice(5512322, 7684834, 1)
slice_chr4: slice(7684834, 10033881, 1)
slice_chr5: slice(10033881, 12510242, 1)
slice_chr6: slice(12510242, 14648397, 1)
slice_chr7: slice(14648397, 16758876, 1)
slice_chr8: slice(16758876, 18817679, 1)
slice_chr9: slice(18817679, 20816493, 1)
slice_chrM: slice(38760292, 38760725, 1)
slice_chrX: slice(37051640, 38619916, 1)
slice_chrY: slice(38619916, 38760292, 1)
zarr_version: 0.1
frac_denominator: num_total_c
frac_min_coverage: 10rdr = ds.reduce_byranges(
ranges=list(regions_dict.values()),
# ranges = [region[["Chromosome", "Start", "End"]] for region in regions_dict.values()],
var=["num_mc", "num_hmc", "num_modc", "num_total_c"]
)
The outcome of reduce_byranges is an xarray.Dataset object which contains summarised methylation information across a genomic range (mean, sum, and CpG count of the range) for each of the variables that we specified in var (typically the number of modified C’s num_mc, or the methylation fraction frac_mc).
# Compute mean methylation levels as mc / total_c
rdr = rdr.assign(
mean_mc = rdr["num_mc_sum"] / rdr["num_total_c_sum"],
mean_hmc = rdr["num_hmc_sum"] / rdr["num_total_c_sum"],
mean_modc = rdr["num_modc_sum"] / rdr["num_total_c_sum"],
)
rdr
<xarray.Dataset> Size: 158MB
Dimensions: (ranges: 323688, sample_id: 1)
Coordinates:
contig (ranges) <U10 13MB 'chr1' 'chr1' ... 'chrY' 'chrY'
start (ranges) int64 3MB 4805787 4805891 ... 89708423
end (ranges) int64 3MB 4806287 4806391 ... 89709423
range_id (ranges) int64 3MB 0 1 2 ... 323685 323686 323687
num_contexts (ranges) int64 3MB 10 10 12 2 0 8 ... 0 0 0 0 0 6
range_length (ranges) int64 3MB 500 500 500 ... 1000 1000 1000
* sample_id (sample_id) <U6 24B 'sample'
Dimensions without coordinates: ranges
Data variables:
num_mc_sum (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_mc_mean (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_mc_cpg_count (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_hmc_sum (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_hmc_mean (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_hmc_cpg_count (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_modc_sum (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_modc_mean (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_modc_cpg_count (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_total_c_sum (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_total_c_mean (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
num_total_c_cpg_count (ranges, sample_id) uint64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
Id (ranges) object 3MB 'ENSMUSG00000025903.14' ......
Gene_id (ranges) object 3MB 'ENSMUSG00000025903.14' ......
Gene_type (ranges) object 3MB 'protein_coding' ... 'prote...
Gene_name (ranges) object 3MB 'Lypla1' ... 'Gm21996'
Level (ranges) object 3MB '2' '2' '2' ... '2' '2' '2'
Mgi_id (ranges) object 3MB 'MGI:1344588' ... 'MGI:5440...
Havana_gene (ranges) object 3MB 'OTTMUSG00000021562.4' ... ...
Tag (ranges) object 3MB 'overlapping_locus' ... ''
Region (ranges) object 3MB 'upstream_2000' ... 'downst...
Strand (ranges) category 324kB <class 'xarray.core.ext...
Parent (ranges) object 3MB nan nan nan ... nan nan nan
Transcript_id (ranges) object 3MB nan nan nan ... nan nan nan
Transcript_type (ranges) object 3MB nan nan nan ... nan nan nan
Transcript_name (ranges) object 3MB nan nan nan ... nan nan nan
Exon_number (ranges) object 3MB nan nan nan ... nan nan nan
Exon_id (ranges) object 3MB nan nan nan ... nan nan nan
Transcript_support_level (ranges) object 3MB nan nan nan ... nan nan nan
Havana_transcript (ranges) object 3MB nan nan nan ... nan nan nan
Protein_id (ranges) object 3MB nan nan nan ... nan nan nan
Ccdsid (ranges) object 3MB nan nan nan ... nan nan nan
Ont (ranges) object 3MB nan nan nan ... nan nan nan
Level_1 (ranges) float64 3MB nan nan nan ... nan nan nan
Ranges_id (ranges) float64 3MB nan nan nan ... nan nan nan
Intron_number (ranges) object 3MB nan nan nan ... nan nan nan
Index (ranges) float64 3MB nan nan nan ... nan nan nan
Bin (ranges) float64 3MB nan nan nan ... nan nan nan
Start_island (ranges) float64 3MB nan nan nan ... nan nan nan
End_island (ranges) float64 3MB nan nan nan ... nan nan nan
Length (ranges) float64 3MB nan nan nan ... nan nan nan
Cpgnum (ranges) float64 3MB nan nan nan ... nan nan nan
Gcnum (ranges) float64 3MB nan nan nan ... nan nan nan
Percpg (ranges) float64 3MB nan nan nan ... nan nan nan
Pergc (ranges) float64 3MB nan nan nan ... nan nan nan
Obsexp (ranges) float64 3MB nan nan nan ... nan nan nan
Type_island (ranges) object 3MB nan nan nan ... nan nan nan
Ranges_ID_island (ranges) float64 3MB nan nan nan ... nan nan nan
Distance (ranges) float64 3MB nan nan nan ... nan nan nan
mean_mc (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
mean_hmc (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>
mean_modc (ranges, sample_id) float64 3MB dask.array<chunksize=(40461, 1), meta=np.ndarray>Add features that require patching#
For each transcript that we select, there are multiple exons and introns. We need to patch them together to have one feature per gene.
def get_mean_across_region(df, var):
sum_var = df[f"{var}_sum"].sum()
sum_total_c = df["num_total_c_sum"].sum()
return sum_var / sum_total_c
def summarise_across_region(rdr, region):
"""
Summarise the methylation data across a region.
Parameters
rdr: pyranges object
region: str
Returns
grouped_df: pd.DataFrame
"""
df = rdr.to_dataframe().reset_index(level="sample_id", drop=True)
grouped = df.groupby("Gene_id")
mean_mc = grouped.apply(get_mean_across_region, var="num_mc")
mean_hmc = grouped.apply(get_mean_across_region, var="num_hmc")
mean_modc = grouped.apply(get_mean_across_region, var="num_modc")
cpg_count = grouped["num_total_c_cpg_count"].sum()
range_length = grouped["range_length"].sum()
gene_name = grouped["Gene_name"].first()
contig = grouped["contig"].first()
grouped_df = pd.DataFrame(
{
"mean_mc": mean_mc,
"mean_hmc": mean_hmc,
"mean_modc": mean_modc,
"cpg_count": cpg_count,
"range_length": range_length,
"Gene_name": gene_name,
"contig": contig,
}
)
grouped_df = grouped_df.reset_index()
grouped_df["Region"] = region
return grouped_df
dict_regions_to_patch = {
"exons": exons,
"introns": introns,
"five_prime_utrs": five_prime_utrs,
"three_prime_utrs": three_prime_utrs,
}
rdr_dict = {}
for region, gr in dict_regions_to_patch.items():
rdr_dict[region] = ds.reduce_byranges(
ranges=gr.unstrand(),
var=["num_mc", "num_hmc", "num_modc", "num_total_c"]
)
dict_df = {}
for region, gr in dict_regions_to_patch.items():
dict_df[region] = summarise_across_region(rdr_dict[region], region)
df_patched_regions = pd.concat(dict_df.values())
df_patched_regions.head()
| Gene_id | mean_mc | mean_hmc | mean_modc | cpg_count | range_length | Gene_name | contig | Region | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | ENSMUSG00000000001.4 | 0.809098 | 0.025972 | 0.848056 | 102 | 2995 | Gnai3 | chr3 | exons |
| 1 | ENSMUSG00000000003.15 | 0.760580 | 0.019347 | 0.793229 | 14 | 681 | Pbsn | chrX | exons |
| 2 | ENSMUSG00000000028.15 | 0.569471 | 0.031448 | 0.608603 | 120 | 1955 | Cdc45 | chr16 | exons |
| 3 | ENSMUSG00000000037.17 | 0.763006 | 0.015735 | 0.789017 | 60 | 2777 | Scml2 | chrX | exons |
| 4 | ENSMUSG00000000049.11 | 0.788420 | 0.031808 | 0.834525 | 56 | 1068 | Apoh | chr11 | exons |
Putting all the features together#
Now for each region we have a mean mC fraction, a mean hmC fraction, a mean modC fraction, as well as the number of CpGs and the length of the region. We can concatenate these results together to have a big dataframe containing all the information we need.
df = rdr.to_dataframe().reset_index(level="sample_id", drop=True)
df["cpg_count"] = df["num_total_c_cpg_count"]
df = df[df_patched_regions.columns].reset_index(drop=True)
df = pd.concat([df, df_patched_regions]).reset_index()
Plots#
We can take this opportunity to look at the distribution of the methylation fraction in all the genomic regions we are interested in.
column_order = [
'upstream_2000', 'upstream_1000', 'upstream_500', 'upstream_250',
'around_tss', 'five_prime_utrs', 'first_exons', 'first_introns',
'exons', 'introns', 'three_prime_utrs', 'genes', 'downstream_1000',
'downstream_2000', 'downstream_3000', 'downstream_4000', 'cpg_islands',
]
g = sns.violinplot(
data=df,
x="Region",
y="mean_mc",
hue="Region",
order=column_order,
cut=0,
palette=biomodal_palette,
saturation=1,
)
g.set_ylabel("Mean 5mC Fraction")
g.set_xticklabels(g.get_xticklabels(), rotation=45, ha="right")
plt.show()
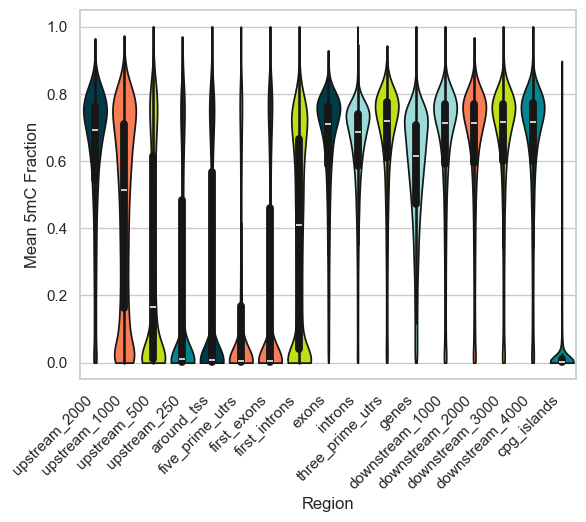
We see that overall, as we approach the TSS from the upstream regions, the methylation fraction starts to decrease. Each of the subregions of the genome have their own methylation levels, and it slowly increases in the downstream regions to go back to mean background level similar to that of the far-away upstream regions. CpG islands have near-zero methylation levels, as expected.
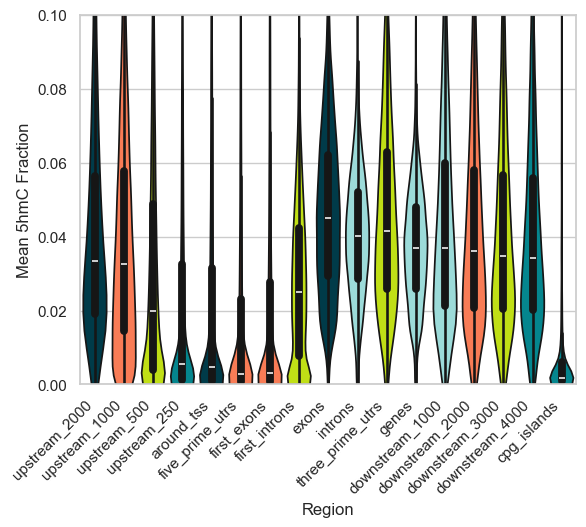
We can do the same plot for the 5hmC levels. Overall the levels of 5hmC in the genome are much lower than that of 5mC, but a similar trend can be observed.
g = sns.violinplot(
data=df,
x="Region",
y="mean_hmc",
hue="Region",
order=column_order,
cut=0,
palette=biomodal_palette,
saturation=1,
)
g.set_ylim(0, 0.1)
g.set_ylabel("Mean 5hmC Fraction")
g.set_xticklabels(g.get_xticklabels(), rotation=45, ha="right")
plt.show()
Create a final feature set#
We want a dataframe containing one line per gene, and where each column is a specific feature (e.g. mean mC fraction at exons, or number of CpGs in the first introns). We want the data to be in this format so that we can easily match it to datasets of gene expression, which will have one expression value per gene.
df_pivot = df.pivot(
index=["Gene_id", "Gene_name", "contig"],
# index = ["Transcript_id", "contig"],
columns="Region",
values=["mean_mc", "mean_hmc", "mean_modc", "cpg_count", "range_length"],
)
df_pivot.head()
| mean_mc | ... | range_length | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | around_tss | cpg_islands | downstream_0 | downstream_1000 | downstream_2000 | downstream_3000 | downstream_4000 | exons | first_exons | first_introns | ... | first_introns | five_prime_utrs | genes | introns | three_prime_utrs | upstream_1000 | upstream_1500 | upstream_2000 | upstream_250 | upstream_500 | ||
| Gene_id | Gene_name | contig | |||||||||||||||||||||
| ENSMUSG00000000001.4 | Gnai3 | chr3 | 0.000474 | 0.000370 | 0.748076 | 0.807040 | 0.749229 | 0.726781 | 0.830068 | 0.809098 | 0.000240 | 0.624894 | ... | 22051.0 | 140.0 | 38866.0 | 13562.0 | 2054.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| ENSMUSG00000000003.15 | Pbsn | chrX | 0.726073 | 0.001538 | 0.696370 | 0.715596 | 0.750000 | 0.644444 | 0.798715 | 0.760580 | 0.726073 | 0.759710 | ... | 5295.0 | 139.0 | 15722.0 | 9532.0 | 235.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| ENSMUSG00000000028.15 | Cdc45 | chr16 | 0.000515 | 0.001563 | 0.780788 | 0.664640 | 0.073536 | 0.012273 | 0.134767 | 0.569471 | 0.001155 | NaN | ... | 15.0 | 311.0 | 31540.0 | 29402.0 | 127.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| ENSMUSG00000000037.17 | Scml2 | chrX | 0.738338 | 0.644383 | 0.731656 | 0.745455 | 0.711864 | 0.762791 | 0.683694 | 0.763006 | 0.741379 | 0.792335 | ... | 34668.0 | NaN | 175688.0 | 138112.0 | NaN | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| ENSMUSG00000000049.11 | Apoh | chr11 | 0.003213 | 0.002256 | 0.786680 | 0.736784 | 0.745746 | 0.733369 | 0.622294 | 0.788420 | 0.826833 | 0.609230 | ... | 51939.0 | 50.0 | 71042.0 | 17921.0 | 100.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
5 rows × 95 columns
df_features = df_pivot.copy()
df_features.columns = [" ".join(col).strip() for col in df_features.columns.values]
features = df_features.columns
# replace white spaces with underscores in features
features = [f.replace(" ", "_") for f in features]
df_features.columns = features
The above dataframe is our feature set, containing a series of features (mean 5mC, mean 5hmC, mean modC, CpG count, and region length) for each of the genomic regions of interest, and for each gene.
Write to file#
Finally, we write this feature file as a pickle file, which preserves the variable type, and is therefore prefered over a more basic text file.
df_features = df_features.reset_index()
df_features.head()
| Gene_id | Gene_name | contig | mean_mc_around_tss | mean_mc_cpg_islands | mean_mc_downstream_0 | mean_mc_downstream_1000 | mean_mc_downstream_2000 | mean_mc_downstream_3000 | mean_mc_downstream_4000 | ... | range_length_first_introns | range_length_five_prime_utrs | range_length_genes | range_length_introns | range_length_three_prime_utrs | range_length_upstream_1000 | range_length_upstream_1500 | range_length_upstream_2000 | range_length_upstream_250 | range_length_upstream_500 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ENSMUSG00000000001.4 | Gnai3 | chr3 | 0.000474 | 0.000370 | 0.748076 | 0.807040 | 0.749229 | 0.726781 | 0.830068 | ... | 22051.0 | 140.0 | 38866.0 | 13562.0 | 2054.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| 1 | ENSMUSG00000000003.15 | Pbsn | chrX | 0.726073 | 0.001538 | 0.696370 | 0.715596 | 0.750000 | 0.644444 | 0.798715 | ... | 5295.0 | 139.0 | 15722.0 | 9532.0 | 235.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| 2 | ENSMUSG00000000028.15 | Cdc45 | chr16 | 0.000515 | 0.001563 | 0.780788 | 0.664640 | 0.073536 | 0.012273 | 0.134767 | ... | 15.0 | 311.0 | 31540.0 | 29402.0 | 127.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| 3 | ENSMUSG00000000037.17 | Scml2 | chrX | 0.738338 | 0.644383 | 0.731656 | 0.745455 | 0.711864 | 0.762791 | 0.683694 | ... | 34668.0 | NaN | 175688.0 | 138112.0 | NaN | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
| 4 | ENSMUSG00000000049.11 | Apoh | chr11 | 0.003213 | 0.002256 | 0.786680 | 0.736784 | 0.745746 | 0.733369 | 0.622294 | ... | 51939.0 | 50.0 | 71042.0 | 17921.0 | 100.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 |
5 rows × 98 columns
# add a column with a boolean depending on whether the transcript is is selected_transcripts or not
df_features["selected_transcript"] = df_features.Gene_id.isin(selected_transcripts.gene_id)
df_features.columns
Index(['Gene_id', 'Gene_name', 'contig', 'mean_mc_around_tss',
'mean_mc_cpg_islands', 'mean_mc_downstream_0',
'mean_mc_downstream_1000', 'mean_mc_downstream_2000',
'mean_mc_downstream_3000', 'mean_mc_downstream_4000', 'mean_mc_exons',
'mean_mc_first_exons', 'mean_mc_first_introns',
'mean_mc_five_prime_utrs', 'mean_mc_genes', 'mean_mc_introns',
'mean_mc_three_prime_utrs', 'mean_mc_upstream_1000',
'mean_mc_upstream_1500', 'mean_mc_upstream_2000',
'mean_mc_upstream_250', 'mean_mc_upstream_500', 'mean_hmc_around_tss',
'mean_hmc_cpg_islands', 'mean_hmc_downstream_0',
'mean_hmc_downstream_1000', 'mean_hmc_downstream_2000',
'mean_hmc_downstream_3000', 'mean_hmc_downstream_4000',
'mean_hmc_exons', 'mean_hmc_first_exons', 'mean_hmc_first_introns',
'mean_hmc_five_prime_utrs', 'mean_hmc_genes', 'mean_hmc_introns',
'mean_hmc_three_prime_utrs', 'mean_hmc_upstream_1000',
'mean_hmc_upstream_1500', 'mean_hmc_upstream_2000',
'mean_hmc_upstream_250', 'mean_hmc_upstream_500',
'mean_modc_around_tss', 'mean_modc_cpg_islands',
'mean_modc_downstream_0', 'mean_modc_downstream_1000',
'mean_modc_downstream_2000', 'mean_modc_downstream_3000',
'mean_modc_downstream_4000', 'mean_modc_exons', 'mean_modc_first_exons',
'mean_modc_first_introns', 'mean_modc_five_prime_utrs',
'mean_modc_genes', 'mean_modc_introns', 'mean_modc_three_prime_utrs',
'mean_modc_upstream_1000', 'mean_modc_upstream_1500',
'mean_modc_upstream_2000', 'mean_modc_upstream_250',
'mean_modc_upstream_500', 'cpg_count_around_tss',
'cpg_count_cpg_islands', 'cpg_count_downstream_0',
'cpg_count_downstream_1000', 'cpg_count_downstream_2000',
'cpg_count_downstream_3000', 'cpg_count_downstream_4000',
'cpg_count_exons', 'cpg_count_first_exons', 'cpg_count_first_introns',
'cpg_count_five_prime_utrs', 'cpg_count_genes', 'cpg_count_introns',
'cpg_count_three_prime_utrs', 'cpg_count_upstream_1000',
'cpg_count_upstream_1500', 'cpg_count_upstream_2000',
'cpg_count_upstream_250', 'cpg_count_upstream_500',
'range_length_around_tss', 'range_length_cpg_islands',
'range_length_downstream_0', 'range_length_downstream_1000',
'range_length_downstream_2000', 'range_length_downstream_3000',
'range_length_downstream_4000', 'range_length_exons',
'range_length_first_exons', 'range_length_first_introns',
'range_length_five_prime_utrs', 'range_length_genes',
'range_length_introns', 'range_length_three_prime_utrs',
'range_length_upstream_1000', 'range_length_upstream_1500',
'range_length_upstream_2000', 'range_length_upstream_250',
'range_length_upstream_500', 'selected_transcript'],
dtype='object')
genes.df.columns
Index(['Chromosome', 'Source', 'Type', 'Start', 'End', 'Score', 'Phase', 'Id',
'Gene_id', 'Gene_type', 'Gene_name', 'Level', 'Mgi_id', 'Havana_gene',
'Tag', 'Ranges_ID', 'Region'],
dtype='object')
df_features = genes.df[["Gene_id", "Chromosome", "Start", "End"]].merge(
df_features, on="Gene_id", how="left",
)
df_features
| Gene_id | Chromosome | Start | End | Gene_name | contig | mean_mc_around_tss | mean_mc_cpg_islands | mean_mc_downstream_0 | mean_mc_downstream_1000 | ... | range_length_five_prime_utrs | range_length_genes | range_length_introns | range_length_three_prime_utrs | range_length_upstream_1000 | range_length_upstream_1500 | range_length_upstream_2000 | range_length_upstream_250 | range_length_upstream_500 | selected_transcript | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ENSMUSG00000025903.14 | chr1 | 4807787 | 4848409 | Lypla1 | chr1 | 0.000753 | 0.000563 | 0.732955 | 0.554563 | ... | 90.0 | 40622.0 | 38089.0 | 1722.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 1 | ENSMUSG00000104217.1 | chr1 | 4807891 | 4886769 | Gm37988 | chr1 | 0.000640 | 0.000563 | 0.700337 | 0.740320 | ... | NaN | 78878.0 | 57461.0 | NaN | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 2 | ENSMUSG00000033813.15 | chr1 | 4857813 | 4897908 | Tcea1 | chr1 | 0.001299 | 0.000860 | 0.816780 | 0.796281 | ... | 99.0 | 40095.0 | 28064.0 | 1540.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 3 | ENSMUSG00000033793.12 | chr1 | 5070017 | 5162528 | Atp6v1h | chr1 | 0.732394 | 0.001306 | 0.820783 | 0.761044 | ... | NaN | 92511.0 | 76589.0 | NaN | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 4 | ENSMUSG00000025905.14 | chr1 | 5588465 | 5606130 | Oprk1 | chr1 | 0.228089 | 0.086325 | 0.772798 | 0.792746 | ... | 183.0 | 17665.0 | 12967.0 | 3346.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 21668 | ENSMUSG00000094739.2 | chrY | 78835720 | 78838055 | Gm20806 | chrY | NaN | 0.648564 | NaN | NaN | ... | 398.0 | 2335.0 | NaN | 138.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 21669 | ENSMUSG00000095867.2 | chrY | 79148788 | 79151121 | Gm20917 | chrY | NaN | 0.648564 | NaN | NaN | ... | 399.0 | 2333.0 | NaN | 138.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 21670 | ENSMUSG00000094660.2 | chrY | 84562571 | 84564906 | Gm21394 | chrY | NaN | 0.648564 | NaN | NaN | ... | 398.0 | 2335.0 | NaN | 138.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 21671 | ENSMUSG00000095650.2 | chrY | 85528516 | 85530907 | Gm20854 | chrY | 0.536585 | 0.648564 | 0.814815 | NaN | ... | NaN | 2391.0 | 6.0 | NaN | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
| 21672 | ENSMUSG00000100608.1 | chrY | 89713423 | 89745531 | Gm21996 | chrY | 0.833333 | 0.648564 | 0.711409 | 0.888889 | ... | 56.0 | 32108.0 | 29823.0 | 203.0 | 500.0 | 500.0 | 500.0 | 250.0 | 250.0 | True |
21673 rows × 102 columns
df_features.to_pickle("features.pickle")
For all 21,673 protein-coding genes in the mm10 genome, this file contains the ID, name, chromosome, start and end, as well as information about each sub-region of the gene (i.e. mean methylation, cpg count, and length of regions such as exons, upstream, etc…). It also contains a transcript ID column to identify the relevant transcript that we kept for each gene.