Working with genomic ranges in modality#
A common pattern when analysing methylation datasets is to perform a series of operations over specified genomic regions. modality has extensive support for working with genomic ranges via pyranges objects from the PyRanges package. In this notebook we showcase some of the working patterns and analyses that are enabled by modality when working with genomic ranges.
Load data#
For the purposes of this demo we can use a duet +modC dataset of Genome in a bottle (GIAB) samples. The data are publicly available and can be loaded using the load_biomodal_dataset() function from the datasets module. This will pull the dataset and load it in to our session as a ContigDataset object.
from modality import load_biomodal_dataset
ds = load_biomodal_dataset("giab")
We will restrict to a single trio in the dataset here for the purposes of the demonstration. Once we have subset the dataset we use assign_fractions method of the ContigDataset to calculate methylation fractions.
sample_selection = ds["family"].values == "Ashkenazi Jewish"
ds = ds.sel(sample_id=sample_selection).copy()
ds.assign_fractions(
numerators="num_modc",
denominator="num_total_c",
min_coverage=10,
inplace=True,
)
Annotation via intervals#
Often we will want to overlay some annotation from external sources onto the genomic regions captured in the ContigDataset. modality provides a series of built-in functions to extract genomic annotations from UCSC and GENECODE. These functions are availble via the modality.annotation module.
Using built-in functions for retrieving annotation#
from modality.annotation import (
get_cpg_islands,
get_cpg_shores,
get_cpg_shelves,
get_exons,
get_five_prime_utrs,
)
We can wrap multiple get_* functions into lists or dictionaries and then use these for downstream analysis:
contigs = [f"chr{i}" for i in range(1, 23)] + ["chrX", "chrY"]
cpg_annotations = [
get_cpg_islands(contigs=contigs),
get_cpg_shelves(contigs=contigs),
get_cpg_shores(contigs=contigs),
]
For example, the first item in the cpg_annotations list here is a pyranges object of CpG islands:
cpg_islands = cpg_annotations[0]
cpg_islands
| Index | Bin | Chromosome | Start | End | Length | Cpgnum | Gcnum | Percpg | Pergc | Obsexp | Type | Ranges_ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 9 | 585 | chr1 | 28735 | 29737 | 1002 | 111 | 731 | 22.2 | 73.0 | 0.85 | cpg_island | 0 |
| 1 | 10 | 586 | chr1 | 135124 | 135563 | 439 | 30 | 295 | 13.7 | 67.2 | 0.64 | cpg_island | 1 |
| 2 | 11 | 586 | chr1 | 199251 | 200121 | 870 | 104 | 643 | 23.9 | 73.9 | 0.89 | cpg_island | 2 |
| 3 | 12 | 587 | chr1 | 368792 | 370063 | 1271 | 99 | 777 | 15.6 | 61.1 | 0.84 | cpg_island | 3 |
| 4 | 13 | 587 | chr1 | 381172 | 382185 | 1013 | 84 | 734 | 16.6 | 72.5 | 0.64 | cpg_island | 4 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 27944 | 189 | 779 | chrY | 25464370 | 25464941 | 571 | 51 | 403 | 17.9 | 70.6 | 0.72 | cpg_island | 27944 |
| 27945 | 190 | 786 | chrY | 26409388 | 26409785 | 397 | 32 | 252 | 16.1 | 63.5 | 0.82 | cpg_island | 27945 |
| 27946 | 191 | 788 | chrY | 26627168 | 26627397 | 229 | 25 | 172 | 21.8 | 75.1 | 0.78 | cpg_island | 27946 |
| 27947 | 192 | 1020 | chrY | 57067645 | 57068034 | 389 | 36 | 257 | 18.5 | 66.1 | 0.85 | cpg_island | 27947 |
| 27948 | 193 | 1021 | chrY | 57203115 | 57203423 | 308 | 29 | 190 | 18.8 | 61.7 | 0.99 | cpg_island | 27948 |
27949 rows × 13 columns
Providing your own set of genomic ranges#
You can also specify you own set of genomic ranges, either by loading in a file, for example a bedfile using pyranges.read_bed or building a ranges object:
import pyranges as pr
custom_ranges = pr.PyRanges(
chromosomes=["chr1", "chr1", "chr1"],
starts=[28735, 135124, 199251],
ends=[29737, 135563, 200121],
)
custom_ranges
| Chromosome | Start | End | |
|---|---|---|---|
| 0 | chr1 | 28735 | 29737 |
| 1 | chr1 | 135124 | 135563 |
| 2 | chr1 | 199251 | 200121 |
Computing over genomic ranges#
Once you have a set of genomic ranges of interest we can use the reduce_byranges() method of the ContigDataset, to summarise methylation information across these ranges. In a bit more detail:
Broadly, from a set of N (chr, start, stop) intervals, applied to a dataset of C (contexts) x S (samples) we transform to an array of (start_ix, stop_ix) windows. When the set of intervals is applied, we select a function (numpy.mean or numpy.sum) that is applied to rows of the selected variable, and applied across axis 0, ie summarizing all values in the window.
Please note: currently we only support mean or sum reduction methods. We hope to address this in future releases
reduced_ds = ds.reduce_byranges(
ranges=cpg_annotations,
var="num_total_c",
)
This method returns an xarray.Dataset with reduced variables now computed over the ranges we specified.
reduced_ds
<xarray.Dataset> Size: 46MB
Dimensions: (ranges: 137687, sample_id: 6)
Coordinates:
contig (ranges) <U20 11MB 'chr1' 'chr1' ... 'chrY'
start (ranges) int64 1MB 28735 135124 ... 57203423
end (ranges) int64 1MB 29737 135563 ... 57205423
range_id (ranges) int64 1MB 0 1 2 ... 137685 137686
num_contexts (ranges) int64 1MB 222 60 208 198 ... 0 0 0
range_length (ranges) int64 1MB 1002 439 ... 2000 2000
group (sample_id) <U22 528B 'CEG1530-EL01-A1200...
* sample_id (sample_id) <U22 528B 'CEG1530-EL01-A1200...
Input DNA Quantity (ng/sample) (sample_id) int64 48B 80 80 80 80 80 80
Protocol Version (sample_id) object 48B '5-Letter v2.4' .....
giab_id (sample_id) object 48B 'HG002' ... 'HG004'
NA_id (sample_id) object 48B 'NA24385' ... 'NA2...
family (sample_id) object 48B 'Ashkenazi Jewish'...
family_status (sample_id) object 48B 'Son' ... 'Mother'
sex (sample_id) object 48B 'Male' ... 'Female'
tech_replicate_number (sample_id) int64 48B 1 1 1 2 2 2
Dimensions without coordinates: ranges
Data variables:
num_total_c_sum (ranges, sample_id) uint64 7MB dask.array<chunksize=(34422, 2), meta=np.ndarray>
num_total_c_mean (ranges, sample_id) float64 7MB dask.array<chunksize=(34422, 2), meta=np.ndarray>
num_total_c_cpg_count (ranges, sample_id) uint64 7MB dask.array<chunksize=(34422, 2), meta=np.ndarray>
Index (ranges) int64 1MB 9 10 11 ... 192 193 193
Bin (ranges) int64 1MB 585 586 586 ... 1021 1021
Length (ranges) int64 1MB 1002 439 870 ... 308 308
Cpgnum (ranges) int64 1MB 111 30 104 ... 36 29 29
Gcnum (ranges) int64 1MB 731 295 643 ... 190 190
Percpg (ranges) float64 1MB 22.2 13.7 ... 18.8 18.8
Pergc (ranges) float64 1MB 73.0 67.2 ... 61.7 61.7
Obsexp (ranges) float64 1MB 0.85 0.64 ... 0.99 0.99
Type (ranges) object 1MB 'cpg_island' ... 'sho...Because we have now substantially reduced the size of the dataset by performing a series of range-based reductions we can now seemlessly integrate with other standard analysis packages such as pandas.
Please Note: This transformation should only be performed when you have used modality to subset or reduce your dataset - transforming to a pandas.DataFrame before you have done so will likely crash your notebook!
reduced_ds_pandas = reduced_ds.to_dataframe()
reduced_ds_pandas
| num_total_c_sum | num_total_c_mean | num_total_c_cpg_count | contig | start | end | range_id | num_contexts | range_length | group | ... | tech_replicate_number | Index | Bin | Length | Cpgnum | Gcnum | Percpg | Pergc | Obsexp | Type | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ranges | sample_id | |||||||||||||||||||||
| 0 | CEG1530-EL01-A1200-004 | 367 | 1.653153 | 222 | chr1 | 28735 | 29737 | 0 | 222 | 1002 | CEG1530-EL01-A1200-004 | ... | 1 | 9 | 585 | 1002 | 111 | 731 | 22.2 | 73.0 | 0.85 | cpg_island |
| CEG1530-EL01-A1200-007 | 303 | 1.364865 | 222 | chr1 | 28735 | 29737 | 0 | 222 | 1002 | CEG1530-EL01-A1200-007 | ... | 1 | 9 | 585 | 1002 | 111 | 731 | 22.2 | 73.0 | 0.85 | cpg_island | |
| CEG1530-EL01-A1200-010 | 314 | 1.414414 | 222 | chr1 | 28735 | 29737 | 0 | 222 | 1002 | CEG1530-EL01-A1200-010 | ... | 1 | 9 | 585 | 1002 | 111 | 731 | 22.2 | 73.0 | 0.85 | cpg_island | |
| CEG1532-EL01-A1200-005 | 324 | 1.459459 | 222 | chr1 | 28735 | 29737 | 0 | 222 | 1002 | CEG1532-EL01-A1200-005 | ... | 2 | 9 | 585 | 1002 | 111 | 731 | 22.2 | 73.0 | 0.85 | cpg_island | |
| CEG1532-EL01-A1200-008 | 401 | 1.806306 | 222 | chr1 | 28735 | 29737 | 0 | 222 | 1002 | CEG1532-EL01-A1200-008 | ... | 2 | 9 | 585 | 1002 | 111 | 731 | 22.2 | 73.0 | 0.85 | cpg_island | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 137686 | CEG1530-EL01-A1200-007 | 0 | NaN | 0 | chrY | 57203423 | 57205423 | 137686 | 0 | 2000 | CEG1530-EL01-A1200-007 | ... | 1 | 193 | 1021 | 308 | 29 | 190 | 18.8 | 61.7 | 0.99 | shores |
| CEG1530-EL01-A1200-010 | 0 | NaN | 0 | chrY | 57203423 | 57205423 | 137686 | 0 | 2000 | CEG1530-EL01-A1200-010 | ... | 1 | 193 | 1021 | 308 | 29 | 190 | 18.8 | 61.7 | 0.99 | shores | |
| CEG1532-EL01-A1200-005 | 0 | NaN | 0 | chrY | 57203423 | 57205423 | 137686 | 0 | 2000 | CEG1532-EL01-A1200-005 | ... | 2 | 193 | 1021 | 308 | 29 | 190 | 18.8 | 61.7 | 0.99 | shores | |
| CEG1532-EL01-A1200-008 | 0 | NaN | 0 | chrY | 57203423 | 57205423 | 137686 | 0 | 2000 | CEG1532-EL01-A1200-008 | ... | 2 | 193 | 1021 | 308 | 29 | 190 | 18.8 | 61.7 | 0.99 | shores | |
| CEG1532-EL01-A1200-011 | 0 | NaN | 0 | chrY | 57203423 | 57205423 | 137686 | 0 | 2000 | CEG1532-EL01-A1200-011 | ... | 2 | 193 | 1021 | 308 | 29 | 190 | 18.8 | 61.7 | 0.99 | shores |
826122 rows × 27 columns
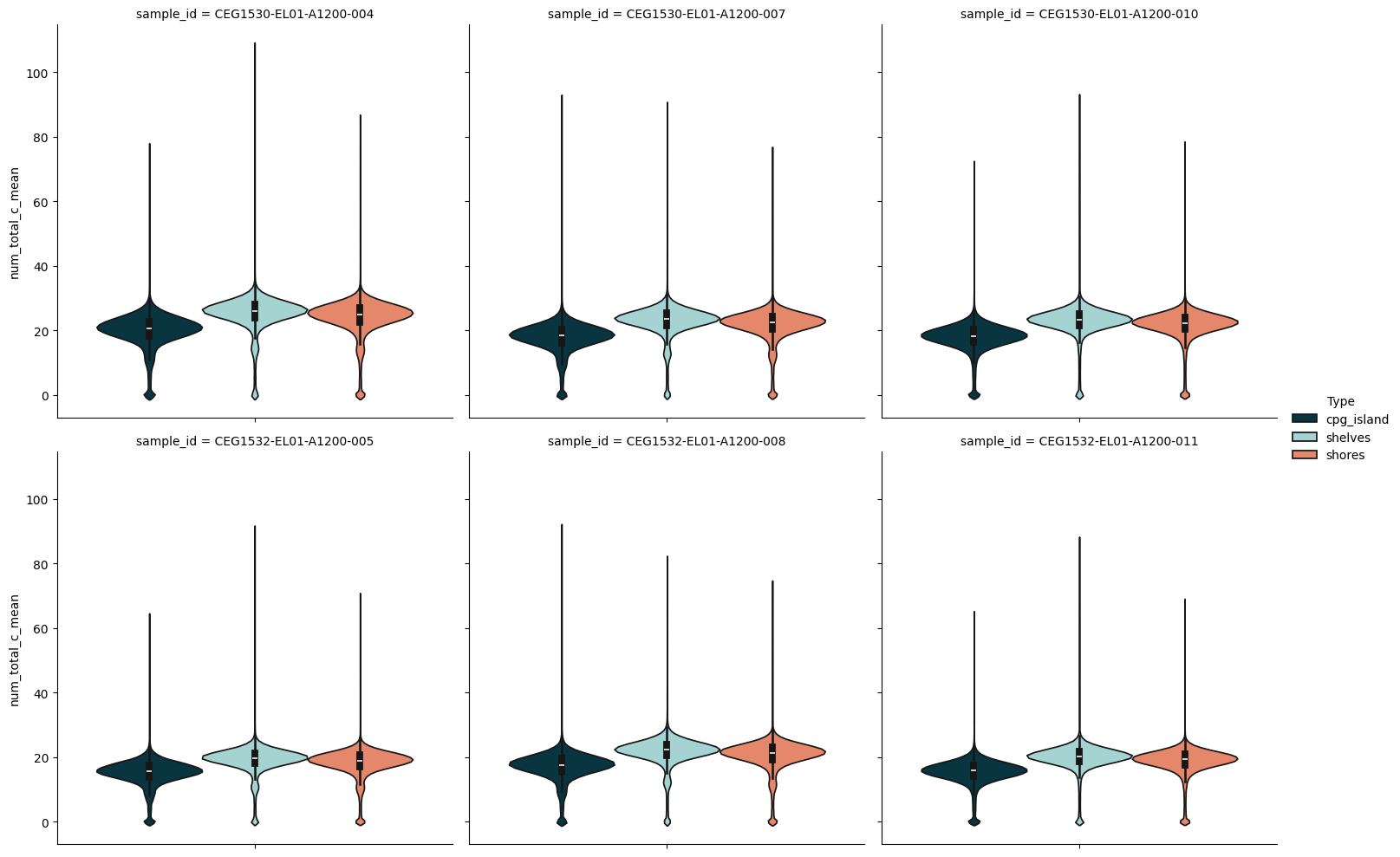
Plotting features from a reduced dataset#
Having provided a set of genomic ranges of interest and then computing methylation information across these ranges and returning a new reduced dataset we can use some of modality's plotting functions to visualise the results. Below we make use of catplot and lmplot from the modality.analysis module.
from modality.analysis import catplot, lmplot
catplot(
reduced_ds,
col="sample_id",
y="num_total_c_mean",
hue="Type",
kind="violin",
col_wrap=3,
)
<seaborn.axisgrid.FacetGrid at 0x37b1bcb10>
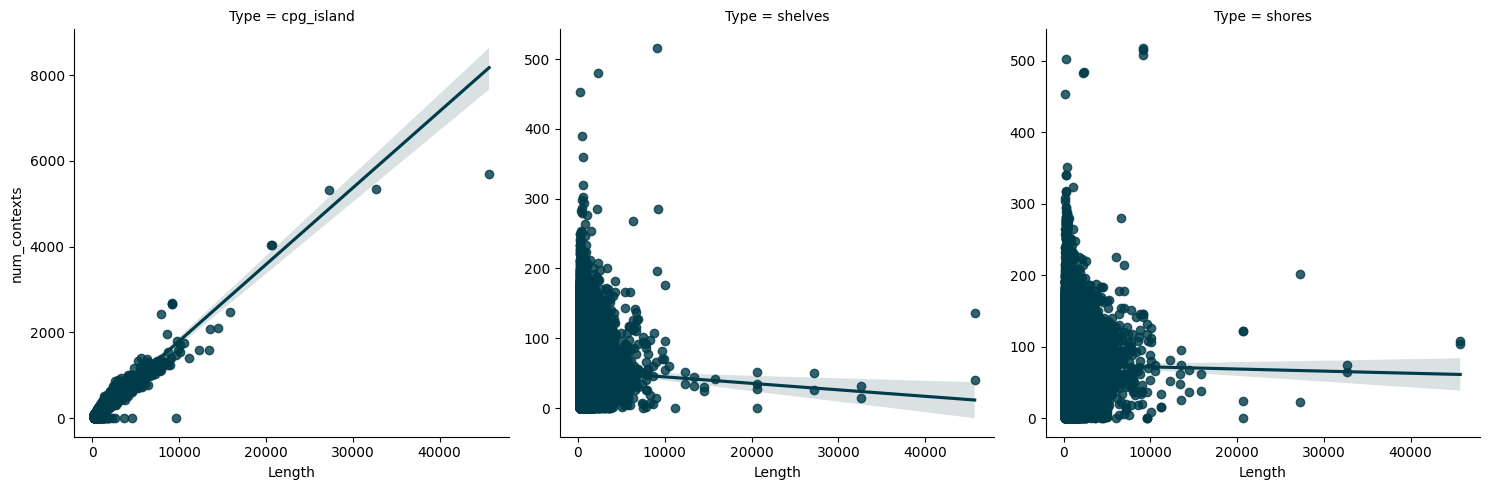
lmplot(reduced_ds, x="Length", y="num_contexts", col="Type", sharey=False)
<seaborn.axisgrid.FacetGrid at 0x38b83ef50>
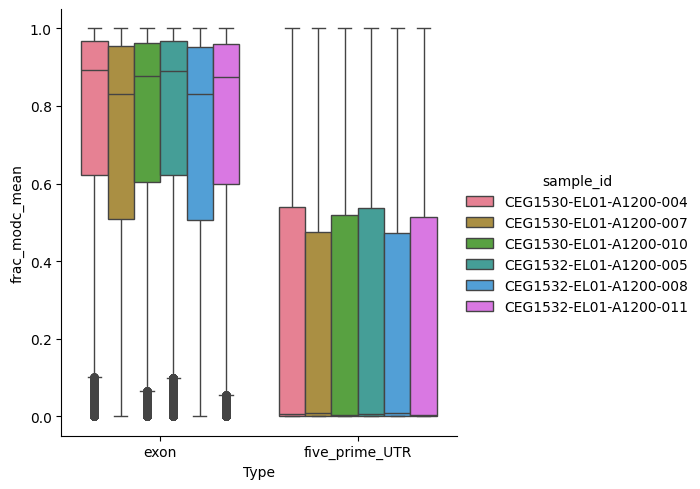
Worked example: Comparing exons to 5-prime UTRs#
As another example, we can quickly reduce the dataset over exons and 5-prime UTRs and compare the methylation fractions between these two classes:
exons_range = get_exons(
contig="chr1",
).unstrand()
five_prime_utr_range = get_five_prime_utrs(
contig="chr1",
).unstrand()
exons_utrs_reduced = ds.reduce_byranges(
ranges=[exons_range, five_prime_utr_range],
var="frac_modc",
)
exons_utrs_reduced
<xarray.Dataset> Size: 31MB
Dimensions: (ranges: 68925, sample_id: 6)
Coordinates:
contig (ranges) <U20 6MB 'chr1' 'chr1' ... 'chr1'
start (ranges) int64 551kB 65418 ... 248859014
end (ranges) int64 551kB 65432 ... 248859084
range_id (ranges) int64 551kB 0 1 2 ... 68923 68924
num_contexts (ranges) int64 551kB 0 0 36 299 ... 0 38 20
range_length (ranges) int64 551kB 14 53 2548 ... 167 70
group (sample_id) <U22 528B 'CEG1530-EL01-A1200...
* sample_id (sample_id) <U22 528B 'CEG1530-EL01-A1200...
Input DNA Quantity (ng/sample) (sample_id) int64 48B 80 80 80 80 80 80
Protocol Version (sample_id) object 48B '5-Letter v2.4' .....
giab_id (sample_id) object 48B 'HG002' ... 'HG004'
NA_id (sample_id) object 48B 'NA24385' ... 'NA2...
family (sample_id) object 48B 'Ashkenazi Jewish'...
family_status (sample_id) object 48B 'Son' ... 'Mother'
sex (sample_id) object 48B 'Male' ... 'Female'
tech_replicate_number (sample_id) int64 48B 1 1 1 2 2 2
Dimensions without coordinates: ranges
Data variables:
frac_modc_sum (ranges, sample_id) float64 3MB dask.array<chunksize=(17232, 3), meta=np.ndarray>
frac_modc_mean (ranges, sample_id) float64 3MB dask.array<chunksize=(17232, 3), meta=np.ndarray>
frac_modc_cpg_count (ranges, sample_id) uint64 3MB dask.array<chunksize=(17232, 3), meta=np.ndarray>
Source (ranges) object 551kB 'HAVANA' ... 'HAVANA'
Type (ranges) object 551kB 'exon' ... 'five_pr...
Score (ranges) object 551kB '.' '.' ... '.' '.'
Phase (ranges) object 551kB '.' '.' ... '.' '.'
Id (ranges) object 551kB 'exon:ENST000006415...
Parent (ranges) object 551kB 'ENST00000641515.2'...
Gene_id (ranges) object 551kB 'ENSG00000186092.7'...
Transcript_id (ranges) object 551kB 'ENST00000641515.2'...
Gene_type (ranges) object 551kB 'protein_coding' .....
Gene_name (ranges) object 551kB 'OR4F5' ... 'ZNF692'
Transcript_type (ranges) object 551kB 'protein_coding' .....
Transcript_name (ranges) object 551kB 'OR4F5-201' ... 'ZN...
Exon_number (ranges) object 551kB '1' '2' ... '1' '1'
Exon_id (ranges) object 551kB 'ENSE00003812156.1'...
Level (ranges) object 551kB '2' '2' ... '2' '2'
Transcript_support_level (ranges) object 551kB '' '' '' ... '1' '1'
Tag (ranges) object 551kB 'RNA_Seq_supported_...
Havana_transcript (ranges) object 551kB 'OTTHUMT00000003223...
Hgnc_id (ranges) object 551kB 'HGNC:14825' ... 'H...
Ont (ranges) object 551kB '' '' '' ... nan nan
Havana_gene (ranges) object 551kB 'OTTHUMG00000001094...
Protein_id (ranges) object 551kB 'ENSP00000493376.2'...
Ccdsid (ranges) object 551kB 'CCDS30547.2' ... '...catplot(
exons_utrs_reduced,
kind='box',
hue='sample_id',
y='frac_modc_mean',
x='Type',
)
<seaborn.axisgrid.FacetGrid at 0x396e3d790>
Subsetting the dataset based on genomic ranges#
As well as reducing the dataset over a series of ranges, modality also supports subsetting the dataset, such that only CpGs that fall within a series of ranges are retained for downstream analysis. This can be very useful to restrict analyses to key regions for example. To do this we can use the subset_byranges() method of the ContigDataset. Below we use this along with a pca analysis (leveraging modality.analysis) to demonstrate this functionality and how one might use it:
from modality.analysis import run_pca, plot_pca_scatter
islands_ds = ds.subset_byranges(
ranges=cpg_islands,
)
islands_ds
<xarray.Dataset> Size: 1GB
Dimensions: (pos: 4315720, sample_id: 6)
Coordinates:
group (sample_id) <U22 528B dask.array<chunksize=(6,), meta=np.ndarray>
* sample_id (sample_id) <U22 528B 'CEG1530-EL01-A1200...
Input DNA Quantity (ng/sample) (sample_id) int64 48B 80 80 80 80 80 80
Protocol Version (sample_id) object 48B '5-Letter v2.4' .....
giab_id (sample_id) object 48B 'HG002' ... 'HG004'
NA_id (sample_id) object 48B 'NA24385' ... 'NA2...
family (sample_id) object 48B 'Ashkenazi Jewish'...
family_status (sample_id) object 48B 'Son' ... 'Mother'
sex (sample_id) object 48B 'Male' ... 'Female'
tech_replicate_number (sample_id) int64 48B 1 1 1 2 2 2
contig (pos) <U20 345MB dask.array<chunksize=(80000,), meta=np.ndarray>
ref_position (pos) uint32 17MB dask.array<chunksize=(80000,), meta=np.ndarray>
strand (pos) <U2 35MB dask.array<chunksize=(80000,), meta=np.ndarray>
trinucleotide_context (pos) uint8 4MB dask.array<chunksize=(80000,), meta=np.ndarray>
Dimensions without coordinates: pos
Data variables:
num_c (pos, sample_id) uint32 104MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
num_modc (pos, sample_id) uint32 104MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
num_n (pos, sample_id) uint32 104MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
num_other (pos, sample_id) uint32 104MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
num_total (pos, sample_id) uint32 104MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
num_total_c (pos, sample_id) uint32 104MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
frac_modc (pos, sample_id) float64 207MB dask.array<chunksize=(80000, 6), meta=np.ndarray>
Attributes:
context: CG
context_encoding: {'0': 'AAA', '1': 'AAC', '10': 'AGA', '100': 'TAA', '...
contigs: ['chr1', 'chr2', 'chr3', 'chr4', 'chr5', 'chr6', 'chr...
coordinate_basis: 0
description: A +modC dataset of human Genome in a bottle (GIAB) sa...
fasta_path: GRCh38Decoy.fa
frac_denominator: num_total_c
frac_min_coverage: 10
input_path: ['CEG1530-EL01-A1200-001.genome.GRCh38Decoy_primary_a...
quant_subtype: modC
ref_name: GRCh38Decoy
sample_ids: ['CEG1530-EL01-A1200-001', 'CEG1530-EL01-A1200-004', ...
slice_chr1: slice(0, 388278, 1)
slice_chr10: slice(2006948, 2207692, 1)
slice_chr11: slice(2207692, 2416334, 1)
slice_chr12: slice(2416334, 2589760, 1)
slice_chr13: slice(2589760, 2683988, 1)
slice_chr14: slice(2683988, 2814352, 1)
slice_chr15: slice(2814352, 2950224, 1)
slice_chr16: slice(2950224, 3173146, 1)
slice_chr17: slice(3173146, 3433468, 1)
slice_chr18: slice(3433468, 3523034, 1)
slice_chr19: slice(3523034, 3841880, 1)
slice_chr2: slice(388278, 671346, 1)
slice_chr20: slice(3841880, 3972924, 1)
slice_chr21: slice(3972924, 4044738, 1)
slice_chr22: slice(4044738, 4168048, 1)
slice_chr3: slice(671346, 852496, 1)
slice_chr4: slice(852496, 1020792, 1)
slice_chr5: slice(1020792, 1214570, 1)
slice_chr6: slice(1214570, 1404772, 1)
slice_chr7: slice(1404772, 1638222, 1)
slice_chr8: slice(1638222, 1813300, 1)
slice_chr9: slice(1813300, 2006948, 1)
slice_chrX: slice(4168048, 4307462, 1)
slice_chrY: slice(4307462, 4315720, 1)
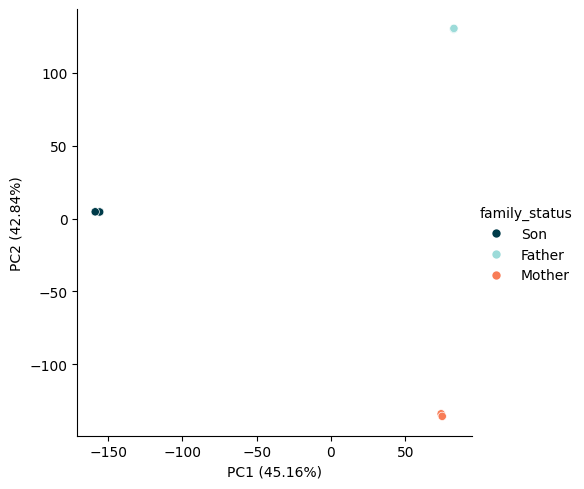
zarr_version: 0.1pca_object, transformed_data = run_pca(
input_array=islands_ds.frac_modc,
)
plot_pca_scatter(
pca_object=pca_object,
pca_result=transformed_data,
hue=islands_ds.family_status,
)
<seaborn.axisgrid.FacetGrid at 0x39bf21f50>