Predict Chromatin Accessibility in ES-E14 Cells, Part II: Training and Evaluating the Model#
This notebook uses duet evoC data to predict chromatin accessibility in mouse embryonic stem cells ES-E14. It uses methylation data in a simple set of genomic regions around the TSS, and learns ATAC-seq values from it, using a public dataset of accessibility. The regions are based on the GENCODE annotations, for the mm10 mouse genome.
For each region, we compute mean 5mC and 5hmC fractions and record the number of CpGs in the region (regardless of whether they are methylated or not). This forms a basis of 3 features per region.
from pathlib import Path
import requests
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import pyranges as pr
import seaborn as sns
import pyBigWig
Note: This notebook requires the additional Machine Learning library XGBoost to train and evaluate models on modality generated data. This notebook was run with XGBoost v2.0.3. You can either install it using your favorite Python package, for example Poetry, or uncomment and run the following cell to install using pip into the current Python virtual environment. Note that the exclamation mark is required in front of the command.
#! pip install xgboost
from scipy.stats import spearmanr
from sklearn.metrics import (
accuracy_score,
mean_absolute_error,
mean_squared_error,
root_mean_squared_error,
r2_score,
roc_auc_score,
f1_score,
)
from sklearn.impute import SimpleImputer
from sklearn.model_selection import GridSearchCV
import xgboost as xgb
from modality.annotation import get_tss_region
sns.set_theme()
sns.set_style("whitegrid")
biomodal_palette = ["#003B49", "#9CDBD9", "#F87C56", "#C0DF16", "#05868E"]
sns.set_palette(biomodal_palette)
Load features from pickle file#
df_features = pd.read_pickle("features_atacseq.pickle")
The above dataframe is our feature set, containing a series of features (mean 5mC, mean 5hmC, and CpG count) for each of the genomic regions of interest, and for each gene.
Load accessibility#
We use the following publicly available ATAC-seq dataset to train our model: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE209527
The ATAC-seq data is given as a bigwig file, representing a track along the genome. We will need to summarise this data, to represent a mean accessibility value near the TSS.
We use the pyBigWig package (pip install pyBigWig), which allows the user to carry out operations directly on the bigwig file.
bigwig_path = "GSE209527_Timeseries_ATACseq_NANOG-FKBP_NT.bw"
if not Path(bigwig_path).exists():
bigwig_source_url = "https://ftp.ncbi.nlm.nih.gov/geo/series/GSE209nnn/GSE209527/suppl/GSE209527%5FTimeseries%5FATACseq%5FNANOG%2DFKBP%5FNT.bw"
response = requests.get(bigwig_source_url)
response.raise_for_status()
with open(bigwig_path, "wb") as file:
for chunk in response.iter_content(chunk_size=8 * 1024):
file.write(chunk)
Extract accessibility around TSS#
Now that we have the obtained the accessibility data from the bigwig file, we want to summarise it over the TSS region.
First, we define a function that takes a chromosome, start, and end position, and uses pyBigWig functionalities to query the relevant region of the bigwig file and return a summary statistic (here, the mean across the region).
In our case, the regions we consider are the regions 500bp on each side of the TSS. We use modality to load the TSS region from GENCODE with the function get_tss_region from the annotation module, which will load the genomic ranges corresponding to the region 500bp each side of the TSS.
def get_region_mean_transcription(region, bigwig_path):
"""
Calculate the region mean transcription using the pyBigWig library.
"""
contig = region.Chromosome
start = region.Start
end = region.End
bigwig = pyBigWig.open(bigwig_path)
positions_mean_transcription = bigwig.values(contig, start, end, numpy=True)
bigwig.close()
weighted_mean_transcription = np.mean(positions_mean_transcription)
return weighted_mean_transcription
span = 1000
start_offset = -500
reference_genome = "mm10"
tss = get_tss_region(
contig=None,
start=None,
end=None,
reference=reference_genome,
protein_coding=True,
filterby=None,
start_offset=start_offset,
span=span,
as_pyranges=True,
)
to_drop = ["Score", "Source", "Phase", "Type", "Level", "Id", "Mgi_id"]
tss = tss.drop(to_drop)
result_df = tss.df
result_df["accessibility"]= result_df.apply(lambda x: get_region_mean_transcription(x, bigwig_path), axis=1)
result_df["Response"] = np.log(result_df["accessibility"])
result_df.head()
| Chromosome | Start | End | Strand | Gene_id | Gene_type | Gene_name | Havana_gene | Tag | Ranges_ID | accessibility | Response | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1 | 4807287 | 4808287 | + | ENSMUSG00000025903.14 | protein_coding | Lypla1 | OTTMUSG00000021562.4 | overlapping_locus | 0 | 26.035425 | 3.259458 |
| 1 | chr1 | 4807391 | 4808391 | + | ENSMUSG00000104217.1 | protein_coding | Gm37988 | OTTMUSG00000050100.1 | overlapping_locus | 1 | 26.583128 | 3.280277 |
| 2 | chr1 | 4857313 | 4858313 | + | ENSMUSG00000033813.15 | protein_coding | Tcea1 | OTTMUSG00000042348.1 | overlapping_locus | 2 | 56.037140 | 4.026015 |
| 3 | chr1 | 5069517 | 5070517 | + | ENSMUSG00000033793.12 | protein_coding | Atp6v1h | OTTMUSG00000050145.9 | 3 | 4.728693 | 1.553649 | |
| 4 | chr1 | 5587965 | 5588965 | + | ENSMUSG00000025905.14 | protein_coding | Oprk1 | OTTMUSG00000034734.3 | 4 | 7.767896 | 2.049999 |
Create final feature file#
In the previous section we have obtained a mean accessibility for the TSS of each protein-codeing gene in the mouse genome. The final piece of work before training a prediction model is to merge this dataset with our 6-base data, which contains methylation data summarised across several sub-regions near the TSS, for the same list of protein-coding genes.
result_df
| Chromosome | Start | End | Strand | Gene_id | Gene_type | Gene_name | Havana_gene | Tag | Ranges_ID | accessibility | Response | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1 | 4807287 | 4808287 | + | ENSMUSG00000025903.14 | protein_coding | Lypla1 | OTTMUSG00000021562.4 | overlapping_locus | 0 | 26.035425 | 3.259458 |
| 1 | chr1 | 4807391 | 4808391 | + | ENSMUSG00000104217.1 | protein_coding | Gm37988 | OTTMUSG00000050100.1 | overlapping_locus | 1 | 26.583128 | 3.280277 |
| 2 | chr1 | 4857313 | 4858313 | + | ENSMUSG00000033813.15 | protein_coding | Tcea1 | OTTMUSG00000042348.1 | overlapping_locus | 2 | 56.037140 | 4.026015 |
| 3 | chr1 | 5069517 | 5070517 | + | ENSMUSG00000033793.12 | protein_coding | Atp6v1h | OTTMUSG00000050145.9 | 3 | 4.728693 | 1.553649 | |
| 4 | chr1 | 5587965 | 5588965 | + | ENSMUSG00000025905.14 | protein_coding | Oprk1 | OTTMUSG00000034734.3 | 4 | 7.767896 | 2.049999 | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 21668 | chrY | 78837555 | 78838555 | - | ENSMUSG00000094739.2 | protein_coding | Gm20806 | OTTMUSG00000046577.2 | 21668 | 0.000000 | -inf | |
| 21669 | chrY | 79150621 | 79151621 | - | ENSMUSG00000095867.2 | protein_coding | Gm20917 | OTTMUSG00000046619.2 | 21669 | 0.000000 | -inf | |
| 21670 | chrY | 84564406 | 84565406 | - | ENSMUSG00000094660.2 | protein_coding | Gm21394 | OTTMUSG00000045415.1 | 21670 | 0.000000 | -inf | |
| 21671 | chrY | 85530407 | 85531407 | - | ENSMUSG00000095650.2 | protein_coding | Gm20854 | OTTMUSG00000042966.1 | 21671 | 0.103704 | -2.266215 | |
| 21672 | chrY | 89745031 | 89746031 | - | ENSMUSG00000100608.1 | protein_coding | Gm21996 | OTTMUSG00000047316.1 | 21672 | 0.103183 | -2.271253 |
21673 rows × 12 columns
df_features_atacseq = pd.merge(
result_df,
df_features,
left_on="Gene_name",
right_on="Gene_name",
suffixes=("_atac", "_6l"),
).sort_values(
by=["Chromosome", "Start"]
)
df_features_atacseq = df_features_atacseq[df_features_atacseq["accessibility"]>0]
df_features_atacseq
| Chromosome | Start | End | Strand | Gene_id_atac | Gene_type | Gene_name | Havana_gene | Tag | Ranges_ID | ... | cpg_count_-1000 | cpg_count_-500 | cpg_count_0 | cpg_count_1000 | cpg_count_500 | range_length_-1000 | range_length_-500 | range_length_0 | range_length_1000 | range_length_500 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 617 | chr1 | 3670997 | 3671997 | - | ENSMUSG00000051951.5 | protein_coding | Xkr4 | OTTMUSG00000026353.2 | 611 | ... | 166.0 | 234.0 | 170.0 | 10.0 | 62.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 618 | chr1 | 4408740 | 4409740 | - | ENSMUSG00000025900.13 | protein_coding | Rp1 | OTTMUSG00000049985.3 | overlapping_locus | 612 | ... | 10.0 | 6.0 | 8.0 | 12.0 | 10.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 |
| 619 | chr1 | 4496853 | 4497853 | - | ENSMUSG00000025902.13 | protein_coding | Sox17 | OTTMUSG00000050014.7 | 613 | ... | 58.0 | 104.0 | 90.0 | 26.0 | 53.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 620 | chr1 | 4785238 | 4786238 | - | ENSMUSG00000033845.13 | protein_coding | Mrpl15 | OTTMUSG00000029329.3 | 614 | ... | 54.0 | 124.0 | 96.0 | 25.0 | 29.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 0 | chr1 | 4807287 | 4808287 | + | ENSMUSG00000025903.14 | protein_coding | Lypla1 | OTTMUSG00000021562.4 | overlapping_locus | 0 | ... | 80.0 | 196.0 | 132.0 | 28.0 | 22.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 21721 | chrY | 85530407 | 85531407 | - | ENSMUSG00000095650.2 | protein_coding | Gm20854 | OTTMUSG00000042966.1 | 21671 | ... | 0.0 | 2.0 | 5.0 | 0.0 | 3.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 21651 | chrY | 87116320 | 87117320 | + | ENSMUSG00000094399.7 | protein_coding | Gm21477 | OTTMUSG00000047083.1 | 21601 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 21653 | chrY | 88052814 | 88053814 | + | ENSMUSG00000101915.1 | protein_coding | Gm28102 | OTTMUSG00000047149.1 | 21603 | ... | 4.0 | 4.0 | 1.0 | 0.0 | 1.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 21654 | chrY | 89052305 | 89053305 | + | ENSMUSG00000102045.1 | protein_coding | Gm21294 | OTTMUSG00000047309.1 | 21604 | ... | 2.0 | 8.0 | 6.0 | 0.0 | 0.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | |
| 21722 | chrY | 89745031 | 89746031 | - | ENSMUSG00000100608.1 | protein_coding | Gm21996 | OTTMUSG00000047316.1 | 21672 | ... | 7.0 | 7.0 | 2.0 | 2.0 | 4.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 | 1000.0 |
20815 rows × 40 columns
Run regressor to predict chromatin accessibility#
We use XGBoost to train a regression model to predict ATAC-seq chromatin accessibility values. The hyperparameters were selected from a grid search.
We split the implementation into several functions which take care of imputing missing data, split the data into testing and training, doing a grid search of hyperparameters if requested, training the model, evaluating the model, and plotting the model versus observations.
By default, we impute the mean of a feature where there is a NaN. NaNs could appear because of lack of coverage in this region. We decided on imputing a mean instead of a 0 as to not conflate the actual 0’s in the features, which correspond to regions containing no methylation.
We split our data between training and testing using held-out chromosomes. By default we use chromosomes 8, 9 and 10, as the test chromosome, and the rest of the genome for training.
Define useful functions#
def impute_missing_values(
data_train,
data_test,
columns_to_keep,
missing_values_strategy="impute_mean",
):
"""
Impute missing values in a dataframe
"""
data_train = data_train.copy()
data_test = data_test.copy()
if missing_values_strategy == "drop":
data_train = data_train[columns_to_keep].dropna()
data_test = data_test[columns_to_keep].dropna()
elif missing_values_strategy == "impute_zero":
data_train = data_train[columns_to_keep].fillna(0)
data_test = data_test[columns_to_keep].fillna(0)
elif missing_values_strategy == "impute_mean":
imputer = SimpleImputer(strategy="mean")
data_train = imputer.fit_transform(data_train[columns_to_keep])
data_test = imputer.transform(data_test[columns_to_keep])
data_train = pd.DataFrame(data_train, columns=columns_to_keep)
data_test = pd.DataFrame(data_test, columns=columns_to_keep)
return data_train, data_test
def select_features(features, mod):
"""
Select features based on the modification type. Only keep the features corresponding to the list `mod`.
E.g. if mod="modc", only keep features related to modC, and discard those related to mC and hmC
"""
if isinstance(mod, str):
mod = [mod]
return [f for f in features if any([m in f for m in mod]) or "cpg_count" in f or "range" in f]
def split_train_test_data(data, features, target, test_contig, missing_values_strategy="impute_mean"):
"""
Split the data into training and testing sets using the specified test contig
"""
if isinstance(test_contig, str):
test_contig = [test_contig]
data_train = data[~data["Chromosome"].isin(test_contig)]
data_test = data[data["Chromosome"].isin(test_contig)]
# data_train = impute_missing_values(data_train, features + [target], missing_values_strategy)
# data_test = impute_missing_values(data_test, features + [target], missing_values_strategy)
data_train, data_test = impute_missing_values(
data_train,
data_test,
features + [target],
missing_values_strategy
)
X_train, y_train = data_train[features], data_train[target]
X_test, y_test = data_test[features], data_test[target]
return X_train, X_test, y_train, y_test
def tune_parameters(X_train, y_train):
"""
Identify the best hyperparameters of the XGBoost regressor using GridSearchCV
"""
param_grid = {
"max_depth": [5, 6, 7, 8],
"n_estimators": [200, 300, 400, 500, 600, 700, 800],
"subsample": [0.4, 0.5, 0.6],
"colsample_bytree": [0.75, 0.8, 0.85, 0.9],
"eta": [0.01, 0.02, 0.03, 0.04, 0.05],
}
regressor = xgb.XGBRegressor(eval_metric="rmsle")
search = GridSearchCV(regressor, param_grid, cv=5, scoring="r2", n_jobs=4).fit(X_train, y_train)
return search.best_params_
def train_model(X_train, y_train, hyperparameters, random_state=1):
"""
Train an XGBoost regressor using the specified hyperparameters
"""
regressor = xgb.XGBRegressor(
random_state=random_state,
**hyperparameters,
)
regressor.fit(X_train, y_train)
return regressor
def evaluate_model(model, X_test, y_test):
"""
Evaluate the performance of the model using the test set
"""
y_pred = model.predict(X_test)
mse = mean_squared_error(y_test, y_pred)
rmse = root_mean_squared_error(y_test, y_pred)
mae = mean_absolute_error(y_test, y_pred)
r2 = r2_score(y_test, y_pred)
spear_r = spearmanr(y_test, y_pred)
return mse, rmse, mae, r2, spear_r
def plot_results(y_test, y_pred, title):
"""
Plot the observed vs predicted accessibility values for the test set
"""
biomodal_palette = ["#003B49", "#9CDBD9", "#F87C56", "#C0DF16", "#05868E"]
plt.plot(y_test, y_pred, ".", ms=4, c=biomodal_palette[0])
# add x=y line
plt.plot([min(y_test), max(y_test)], [min(y_test), max(y_test)], "--", color=biomodal_palette[2])
plt.xlabel("Observed log(accessibility)")
plt.ylabel("Predicted log(accessibility)")
plt.title(title)
plt.xlim(min(y_test)-2, max(y_test)+2)
plt.ylim(min(y_pred)-2, max(y_pred)+2)
plt.grid(True)
plt.show()
def run_regressor(
data,
features,
mod,
target,
hyperparameters,
random_state=1,
missing_values_strategy="impute_mean",
test_contig=None,
find_optimal_parameters=False
):
"""
Run the XGBoost regressor using the specified data and parameters
"""
features = select_features(features, mod)
X_train, X_test, y_train, y_test = split_train_test_data(
data,
features,
target,
test_contig,
missing_values_strategy,
)
if find_optimal_parameters:
best_params = tune_parameters(X_train, y_train)
hyperparameters.update(best_params)
model = train_model(X_train, y_train, hyperparameters, random_state)
mse, rmse, mae, r2, spear_r = evaluate_model(model, X_test, y_test)
df_metrics = pd.DataFrame({
"mse": [mse],
"rmse": [rmse],
"mae": [mae],
"r2": [r2],
"spearman": [spear_r[0]],
})
y_pred = model.predict(X_test)
return model, df_metrics, y_test, y_pred
Run the model#
Let’s run the model in the case where we want to train a chromatin accessibility predictor using mC and hmC methylation levels.
First we need to pass a dictionary of hyperparameters for the XGBoost model, which we obtained through a grid search.
model_hyperparameters = {
'n_estimators': 500,
'max_depth': 6,
'colsample_bytree': 0.85,
'eta': 0.02,
'subsample': 0.6,
}
Features are all the columns in df_features_atacseq that start with “mean” or “count”. Note that in this work, all the genomic ranges have the same length of 1000bp, so we do not include the region length as a feature of our model.
features = [
c for c in df_features_atacseq.columns if c.startswith("mean")
or c.startswith("cpg_count")
]
features
['mean_mc_-1000',
'mean_mc_-500',
'mean_mc_0',
'mean_mc_1000',
'mean_mc_500',
'mean_hmc_-1000',
'mean_hmc_-500',
'mean_hmc_0',
'mean_hmc_1000',
'mean_hmc_500',
'mean_modc_-1000',
'mean_modc_-500',
'mean_modc_0',
'mean_modc_1000',
'mean_modc_500',
'cpg_count_-1000',
'cpg_count_-500',
'cpg_count_0',
'cpg_count_1000',
'cpg_count_500']
model, df_metrics, y_test, y_pred = run_regressor(
data=df_features_atacseq,
features=features,
mod = ["mc", "hmc"],
target="Response",
hyperparameters=model_hyperparameters,
random_state=0,
test_contig=["chr8", "chr9", "chr10"],
missing_values_strategy="impute_mean",
find_optimal_parameters=False,
)
The model performs well, with an r^2 of 0.82 and a Spearman R correlation of 0.92.
df_metrics
| mse | rmse | mae | r2 | spearman | |
|---|---|---|---|---|---|
| 0 | 0.379055 | 0.615675 | 0.46464 | 0.8808 | 0.888941 |
Let’s plot the predictions of the models versus the actual data on the held chromosome 8.
plot_results(
y_test,
y_pred,
f"ATAC-seq - R^2={df_metrics.r2.values[0]:.2f}, Spearman R={df_metrics.spearman.values[0]:.2f}"
)
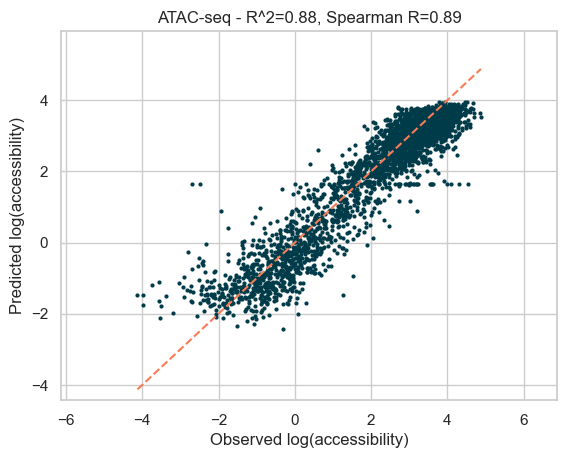
In the above model training, we set a pseudo-random state for reproducibility, but we need to ensure our accuracy estimates are robust to alternative starting random states. Below we train the data again with different pseudo-random states, to get an average value for r^2 and Spearman R.
df_regressor = pd.DataFrame(columns=["mse", "rmse", "mae", "r2", "spearman"])
for k in range(0,20):
model, df_metrics, _, _ = run_regressor(
data=df_features_atacseq,
features=features,
mod = ["mc", "hmc"],
target="Response",
hyperparameters=model_hyperparameters,
random_state=k,
test_contig=["chr8", "chr9", "chr10"],
missing_values_strategy="impute_mean",
find_optimal_parameters=False,
)
df_regressor = pd.concat([df_regressor, df_metrics], ignore_index=True)
df_regressor = df_regressor.reset_index(drop=True)
The average r^2 is at 0.87 and the average Spearman R is at 0.88.
df_regressor[["r2", "spearman"]].describe()
| r2 | spearman | |
|---|---|---|
| count | 20.000000 | 20.000000 |
| mean | 0.880447 | 0.888616 |
| std | 0.000427 | 0.000293 |
| min | 0.879774 | 0.887959 |
| 25% | 0.880155 | 0.888436 |
| 50% | 0.880378 | 0.888534 |
| 75% | 0.880823 | 0.888796 |
| max | 0.881227 | 0.889219 |
Classifier#
Depending on the biological question you are trying to answer, having a binary classifier may be more than sufficient and you may not need to train a regressor. We can also train such a classifier using XGBoost. The classifier can be binary (i.e. two categories: high accessibility and low accessibility), or have more categories (e.g., we could classify genes as having low accessibility, moderate accessibility, or high accessibility). Having more classes will likely result in a drop in the accuracy of the classifier (since it will have more opportunities to misclassify).
def train_classifier_model(X_train, y_train, hyperparameters, random_state=1):
"""
Train an XGBoost classifier using the specified hyperparameters
"""
classifier = xgb.XGBClassifier(
random_state=random_state,
**hyperparameters,
)
classifier.fit(X_train, y_train)
return classifier
def evaluate_classifier(model, X_test, y_test):
"""
Evaluate the performance of the classifier using the test set
"""
y_pred = model.predict(X_test)
y_pred_proba = model.predict_proba(X_test)
accuracy = accuracy_score(y_test, y_pred)
# f1 score
f1 = f1_score(y_test, y_pred, average="macro")
# calculate AUC
if len(np.unique(y_test)) == 2:
auc = roc_auc_score(y_test, y_pred_proba[:, 1])
else:
auc = roc_auc_score(y_test, y_pred_proba, multi_class="ovr", average="macro")
return accuracy, f1, auc
def run_classifier(
data,
features,
mod,
target,
hyperparameters,
random_state=1,
missing_values_strategy="impute_mean",
test_contig=None,
):
"""
Run an xgboost classifier
"""
features = select_features(features, mod)
X_train, X_test, y_train, y_test = split_train_test_data(
data,
features,
target,
test_contig,
missing_values_strategy,
)
classifier = train_classifier_model(X_train, y_train, hyperparameters, random_state)
accuracy, f1, auc = evaluate_classifier(classifier, X_test, y_test)
df_metrics = pd.DataFrame(
{
"number_of_categories": [len(np.unique(y_train))],
"accuracy": [accuracy],
"macro_f1": [f1],
"auc": [auc],
}
)
return classifier, df_metrics
Loop over the number of categories and evaluate the metrics of the classifier.
df_classifier = pd.DataFrame(columns=["number_of_categories", "accuracy", "macro_f1", "auc"])
number_of_categories = np.arange(2, 4, 1)
for n in number_of_categories:
# create n categories based on the response values
labels = [k for k in range(n)]
df_features_atacseq["category"] = pd.cut(
df_features_atacseq["Response"], bins=n, labels=labels
)
# run the classifier
c, df_metrics = run_classifier(
data=df_features_atacseq,
features=features,
mod=["mc", "hmc"],
target="category",
hyperparameters=model_hyperparameters,
random_state=1,
test_contig=["chr8", "chr9", "chr10"],
)
df_classifier = pd.concat([df_classifier, df_metrics], ignore_index=True)
A binary classifier achieves a good accuracy of 94% and AUC of 0.92. As the number of categories increase, the accuracy and F1 score decrease, since the classifier has more opportunities to misclassify. However, with three categories (i.e., low accessibility, moderate accessibility, or high accessibility) we still retain a robust accuracy of 90%. Note that we report the macro-F1 score, where the F1 score is calculated for each class independently, and the unweighted average of these scores is returned.
df_classifier
| number_of_categories | accuracy | macro_f1 | auc | |
|---|---|---|---|---|
| 0 | 2 | 0.948369 | 0.696455 | 0.956680 |
| 1 | 3 | 0.954387 | 0.626521 | 0.981808 |
Feature importance#
XGboost feature importance#
Let’s look at the feature importance of our model, first using XGBoost’s own model.feature_importances_ functionality. This measures the improvement in accuracy brought by a feature to the branches it is involved in. It is a measure of the contribution of a feature to the model.
df_features_importance = pd.DataFrame(
{
"feature": select_features(features, ["mc", "hmc"]),
"importance": model.feature_importances_,
}
)
df_features_importance.sort_values("importance", ascending=False).head(10)
| feature | importance | |
|---|---|---|
| 1 | mean_mc_-500 | 0.650006 |
| 6 | mean_hmc_-500 | 0.089454 |
| 2 | mean_mc_0 | 0.066663 |
| 11 | cpg_count_-500 | 0.052648 |
| 5 | mean_hmc_-1000 | 0.032733 |
| 7 | mean_hmc_0 | 0.017024 |
| 10 | cpg_count_-1000 | 0.014098 |
| 12 | cpg_count_0 | 0.014047 |
| 14 | cpg_count_500 | 0.011358 |
| 0 | mean_mc_-1000 | 0.010187 |
fig, ax = plt.subplots(figsize=(14, 6))
sns.barplot(
x="feature",
y="importance",
data=df_features_importance.sort_values("importance", ascending=False),
)
plt.xticks(rotation=60, ha="right")
plt.show()
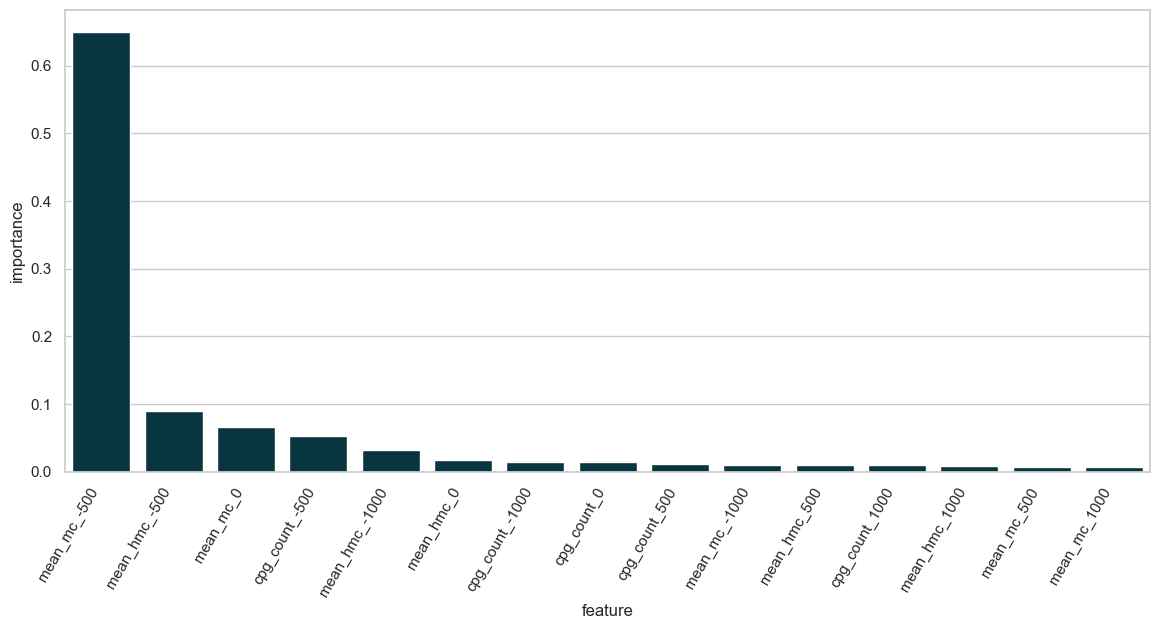
The region extending from 500 base pairs upstream to 500 base pairs downstream of the transcription start site (TSS) is the most significant contributor to predicting chromatin accessibility as measured by ATAC-seq.
Region importance#
Rather than assessing the importance of each feature individually, we aim to gain biological insights into which genomic regions contribute the most to the model’s performance (as measured by their R^2). To achieve this, we ran our regressor on each region separately and recorded their R^2 score. This approach allows us to identify specific regions that have the greatest impact on chromatin accessibility, providing a clearer understanding of the underlying biological mechanisms and potentially highlighting key regulatory elements.
N_runs = 10
df_regressor = pd.DataFrame(columns=["mse", "rmse", "mae", "r2", "spearman", "Region"])
for region in [-1000, -500, 0, 500, 1000]:
columns = [f"mean_mc_{region}", f"mean_hmc_{region}", f"cpg_count_{region}"]
all_columns = columns + ["Response", "Chromosome"]
df_one_region = df_features_atacseq[all_columns]
for k in range(0, N_runs):
_, df_metrics, _, _ = run_regressor(
data=df_one_region,
features=columns,
mod=["mc", "hmc"],
target="Response",
hyperparameters=model_hyperparameters,
random_state=k,
test_contig=["chr8", "chr9", "chr10"],
missing_values_strategy="impute_mean",
find_optimal_parameters=False,
)
df_metrics["Region"] = region
df_regressor = pd.concat([df_regressor, df_metrics], ignore_index=True)
Let’s see if there are regions where hmC contributes significantly to the performance of the model. In order to visualise this, we repeat the experiment above, this time looking only at region importance for a model with mC but no hmC.
N_runs = 10
df_regressor_mc_only = pd.DataFrame(columns=["mse", "rmse", "mae", "r2", "spearman", "Region"])
for region in [-1000, -500, 0, 500, 1000]:
columns = [f"mean_mc_{region}", f"cpg_count_{region}"]
all_columns = columns + ["Response", "Chromosome"]
df_one_region = df_features_atacseq[all_columns]
for k in range(0, N_runs):
_, df_metrics, _, _ = run_regressor(
data=df_one_region,
features=columns,
mod="mc",
target="Response",
hyperparameters=model_hyperparameters,
random_state=k,
test_contig=["chr8", "chr9", "chr10"],
missing_values_strategy="impute_mean",
find_optimal_parameters=False,
)
df_metrics["Region"] = region
df_regressor_mc_only = pd.concat([df_regressor_mc_only, df_metrics], ignore_index=True)
Let’s merge the two experiments and plot the results.
df_regressor_mc_only["features"] = "mc_only"
df_regressor["features"] = "mc+hmc"
df_regions = pd.concat([df_regressor, df_regressor_mc_only])
sns.catplot(
data=df_regions,
x="Region",
y="r2",
hue="features",
kind="bar",
palette=biomodal_palette,
height=5,
aspect=2,
)
plt.xticks(rotation=45, ha="right")
plt.xlabel("Region relative to the TSS")
plt.ylabel(r"$R^2$")
plt.show()
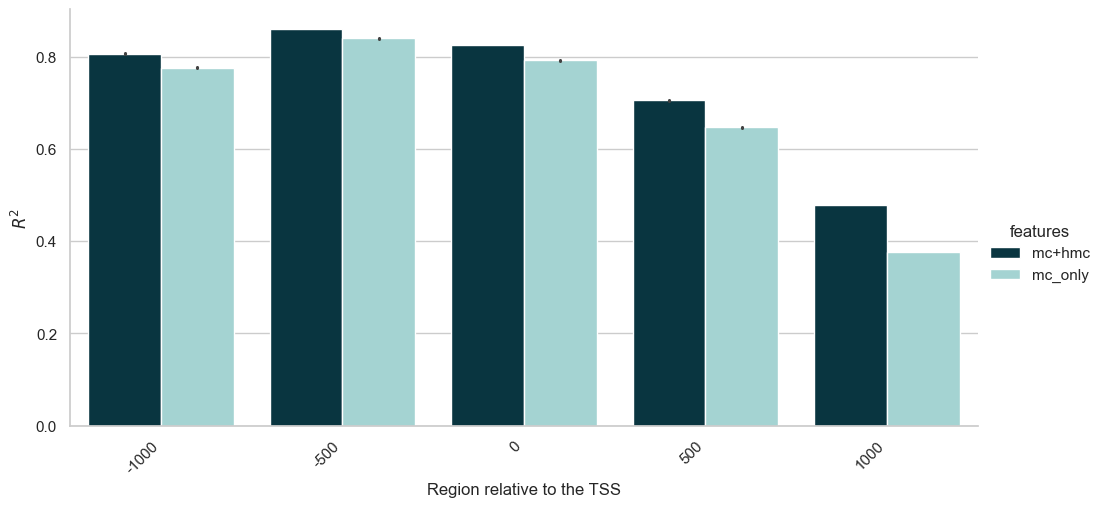
From the plot above we can draw a few conclusions:
The results show that the model performs better when using both mC and hmC features compared to using only mC features.
As we get further from the gene in either direction (upstream or downstream), the contribution of the feature to accessibility decreases.
In summary, this series of notebooks illustrates how modality can help constructing robust machine learning models. It achieves this by condensing methylation data across genomic ranges specified by external annotations, and efficiently grouping them together to generate feature sets, thereby facilitating the model-building process.
This notebook also highlights that the methylation signal obtained from duet evoC data contains sufficient information to make robust chromatin accessibility predictions.